En transposant la loi d’Ohm à l’hémodynamique, on obtient l’équation utilisée pour calculer les résistances artérielles : RAP = (PAPm – POG) / DC. Si l’on extrait la pression artérielle pulmonaire de cette équation, on trouve : PAP ≈ POG + (DC • RAP). La pression de l’AP dépend donc de trois facteurs physiopathologiques [28].
- Pression de l'OG (ou PAPO); hypertension pulmonaire postcapillaire (cardiopathies gauches, maladie veino-occlusive).
- Débit cardiaque; surcharge de volume (shunt gauche – droit, insuffisance tricupidienne sévère).
- Résistance artérielle pulmonaire (résistance statique représentant 75% de la postcharge du VD); hypertension artérielle précapillaire (HTAP primaire, embolie pulmonaire, BPCO, hypoxie). L'impédance (résistance due à la pulsatilité de l’arbre pulmonaire) n'est pas mesurée en clinique, mais elle représente 25% de la postcharge du VD.
Définition et étiologies
L'hypertension pulmonaire (HTP) est définie par une pression moyenne (PAPm) supérieure à 25 mmHg au repos et des RAP > 240 dynes•s•cm-5 ou > 3 U Wood (valeur normale : 60-150 dynes•s•cm-5, < 2 U Wood) [15,38,54]. L’élévation de la PAPm > 30 mmHg à l’effort n’est plus retenue dans la définition de l’HTAP, car cette valeur peut être atteinte chez l’individu normal. Avec l’âge, la PAP s’élève de 1 mmHg par tranche de 10 ans [28]. Elle augmente aussi parallèlement au BMI.
Conceptuellement, l’abbréviation HTAP est réservée à l’hypertension artérielle pulmonaire liée à une augmentation des résistances artériolaires, alors que le terme HTP recouvre l’ensemble des lésions qui entraînent une élévation de la PAP (lésions pulmonaires tissulaires, hypertension veineuse pulmonaire, etc). Dans l’usage courant, on ne fait souvent pas la différence. L’étiologie de l’HTP relève de 5 groupes de causes, selon la classification de l’OMS (Tableau 5.8) [25,53,54].
- 1 - Hypertension artérielle pulmonaire (HTAP) proprement dite :
- HTAP primaire (idiopathique familiale), la prévalence de l’HTAP primaire est de 15 cas (France) à 25 cas (Suisse) par million d’habitants (5% des cas d’HTP) [58].
- HTAP des cardiopathies congénitales (10% des cas adultes).
- HTAP secondaire des médicaments (anorexigènes comme l’aminorex, la fenfluramine ou le benfluorex, cocaïne, amphétamines, imatinib, interféron), à des maladies (HIV, hypertension portale, schistostomiase, sclérodermie), à l’obésité ou à l’âge ; vu sa fréquence (200 millions d’humains atteints), la schistostomiase est probablement la principale cause d’HTAP dans le monde.
- 1’ - Maladie pulmonaire veino-occlusive.
- 1’’ - HTAP persistante du nouveau-né (2 :1'000 bébés).
- 2 - Hypertension pulmonaire (HTP) postcapillaire (P télédiast VG > 18 mmHg, PAPO > 15 mmHg, mais RAP < 3 U Wood et gradient transpulmonaire < 12 mmHg) : défaillance systolique ou insuffisance diastolique restrictive du VG, valvulopathie mitrale. C’est la cause la plus fréquente d’HTP chez l’adulte (65% des cas) [13,59].
- 3 - HTAP associée à l’hypoxie alvéolaire : BPCO, emphysème, SDRA, apnée du sommeil (SAS), hypoxie d’altitude, PEEP excessive. L’élévation de la PAP est en général modérée (PAPm 25-35 mmHg).
- 4 - HTAP liée à la maladie thrombo-embolique pulmonaire chronique ; 4% des embolies pulmonaires aiguës se soldent par une non-résorption des thrombi.
- 5 - HTAP d’origine multifactorielle non éclaircie (sarcoïdose, histiocytose X, maladie de Gaucher, anémie hémolytique, maladies myéloprolifératives, insuffisance rénale dialysée).

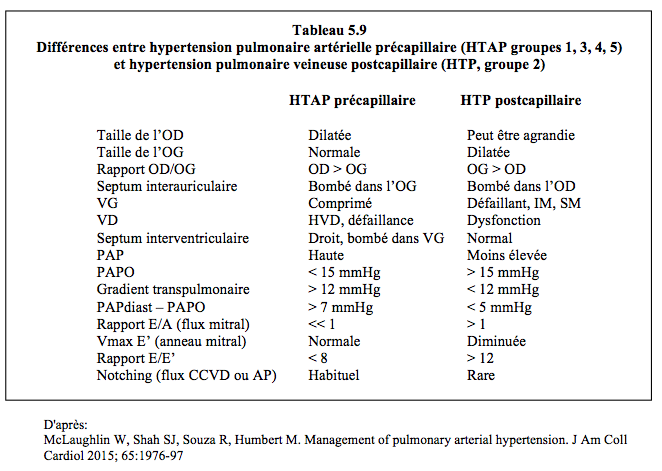
Dans les groupes 1, 3, 4 et 5, l’HTAP est précapillaire ; la POG et la PAPO sont normales (PAPO < 15 mmHg), et le gradient transpulmonaire (GTP = PAPm – POG, ou PAPm - PAPO) est supérieur à 12 mmHg ; les RAP sont élevées (> 240 dynes•s•cm-5 ou > 3 U Wood). Dans le groupe 2, l’HTP est primairement postcapillaire : POG élevée et PAPO > 16 mmHg, RAP et gradient transpulmonaire normaux (GTP ≤ 12 mmHg) ; le gradient entre la PAPdiast et la PAPO est < 5 mmHg (Tableau 5.9) (Figure 5.123).
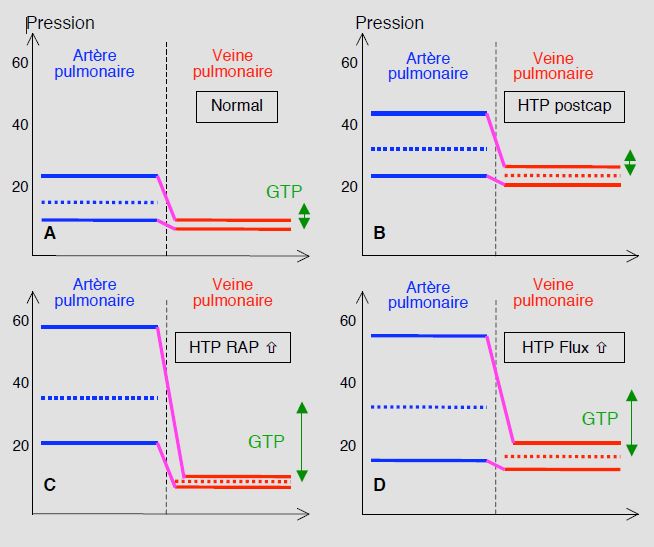
Figure 5.123: Classification schématique des différents mécanismes de l'hypertension pulmonaire (HTP) [44]. A: situation normale; le gradient transpulmonaire (GTP, double flèche verte) est < 12 mmHg. B: HTP postcapillaire ou passive; la pression veineuse est élevée, ce qui entraine une élévation secondaire de la PAP mais le gradient transpulmonaire reste normal. C: HTP précapillaire ou résistive; le mécanisme primaire est une élévation des RAP; la pression veineuse est normale et le GTP est très élevé (> 12 mmHg). D: HTP hyperkinétique secondaire à une augmentation du flux sanguin pulmonaire (shunt G-D); la PAP est élevée, mais la pression veineuse est légèrement supérieure à la norme, ce qui réduit un peu l'augmentation du GTP.
Les nouveaux concepts issus de la biochimie et de la génétique tendent à regrouper les mécanismes étiologiques de l’HTAP sous cinq rubriques différentes [1].
- Une dysfonction endothéliale crée un déséquilibre favorisant la vasoconstriction, la thrombose et la mitogenèse.
- Un polymorphisme génétique modifie l’expression des gènes commandant certains canaux ioniques comme les canaux potassiques dépendant du voltage (KV) ; le non-fonctionnement des canaux KV entraîne une dépolarisation de la membrane qui augmente la concentration cytoplasmique de K+ et de Ca2+, d’où activation de la contraction de la musculature lisse. Les canaux KV sont également inhibés par l’hypoxie ; leur perte conduit à la vasoconstriction pulmonaire hypoxique (voir ci-dessous Figure 5.126).
- Une prolifération cellulaire excessive, une baisse de l’apoptose et un métabolisme dévié vers la glycolyse (excès de pyruvate-déhydrogénase-kinase qui bloque le cycle de Krebs) dans les cellules musculaires lisses, les fibroblastes et l’endothélium suggèrent des mécanismes analogues à ceux de la cellule cancéreuse.
- Une vasoconstriction réfractaire peut s’installer par suractivation de la phospholipase C ; elle ne réagit plus aux vasodilatateurs (anticalciques, nitroglycérine). Ainsi, seuls 20% des patients souffrant d’HTAP répondent aux bloqueurs calciques.
- L’état fonctionnel du VD est l’élément déterminant dans le pronostic de l’HTAP. L’augmentation de postcharge induit une hypertrophie ventriculaire droite (HVD), caractérisée par une surexpression de la phosphodiestérase-5, qui métabolise le GMPc, et un excès de pyruvate-déhydrogénase-kinase, qui bloque le cycle de Krebs. Bien qu’hypertrophié, le VD est donc métaboliquement dysfonctionnel.
L’HTAP chronique présente une progression dans les lésions anatomo-pathologiques, typiques d’une panvasculopathie (Figure 5.124) [45].
- Hyperplasie intimale, épaississement des cellules endothéliales.
- Hypertrophie de la média et muscularisation réversible des portions terminales de l’arbre pulmonaire.
- Prolifération des fibroblastes adventitiels.
- Infiltration de cellules inflammatoires.
- Occlusion progressive et thrombose des petits vaisseaux ; aux phases avancées, l’arbre pulmonaire est constellé de thrombus muraux, d’où l’importance d’anticoaguler ces malades [5].
- Lésions plexiformes d’angioprolifération dans l’HTAP idiopathique oblitérant les artérioles ; cette lésion ne se rencontre pas dans l’HTP postcapillaire ni dans l’hypoxie pulmonaire.
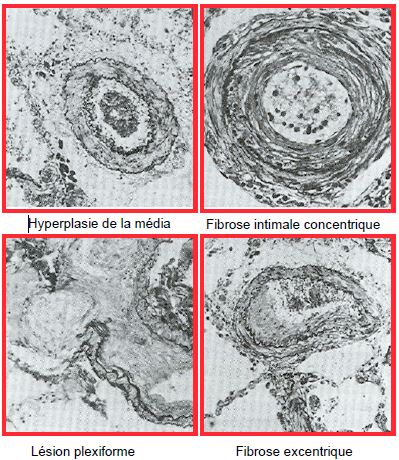
Figure 5.124: Coupes histologiques de l’évolution anatomo-pathologique de l’hypertension pulmonaire chronique [45].
La distribution des lésions est très hétérogène. La pathologie épargne les veines, la circulation bronchique, le réseau systémique et les bronches. Au stade terminal, l’HTAP sévère est fixée (PAPm > 55 mmHg, RAP > 800 dynes•s•cm-5) et n’est plus modulable ; c’est le syndrome d’Eisenmenger [43].
L’HTP liée aux pathologies du cœur gauche (groupe 2) est la catégorie de loin la plus fréquente d’hypertension pulmonaire ; la moyenne d’âge y est plus élevée. Dans ce groupe, l’HTP est primairement postcapillaire : POG et PAPO élevées (> 15 mmHg), RAP et gradient transpulmonaire bas (GTP < 15 mmHg). Le gradient entre la PAPdiast et la PAPO est < 5 mmHg; il est plus sensible et plus spécifique que le GTP [13,16,59]. Cette élévation passive de la PAP est liée à l’augmentation de pression dans l’OG et dans les veines pulmonaires ; elle s'accompagne d'un exsudat intersticiel et d'un dysfonctionnement des pompes Na+-K+ qui extraient l'eau des alvéoles [22]. Elle est réversible si la lésion gauche est corrigée (chirurgie de la valve mitrale, correction de la défaillance du VG). Cependant, cette HTP, qui induit aussi une dysfonction endothéliale avec altération de la production de NO et d’endothéline, est le plus souvent accompagnée d’une vasoconstriction artériolaire réactionnelle (HTAP) qui augmente la PAP hors de proportion avec l’élévation de la pression veineuse pulmonaire (gradient transpulmonaire > 12 mmHg dans 45% des cas de PAPO > 15 mmHg). La finalité de cette vasoconstriction due au remodelage artériolaire est probablement de diminuer l’engorgement pulmonaire en freinant le débit : l’administration de vasodilatateurs pulmonaires (NO, époprosténol, bosentan) dans cette situation tend à augmenter la congestion capillaire et la POG et n’améliore pas le débit cardiaque ; elle peut même conduire à l’OAP [21,59]. Cette combinaison d'HTP pré- et post-capillaire est caractérisée par un gradient diastolique (PAPdiast – PAPO) > 7 mmHg, qui est lui-même un prédicteur indépendant de mortalité [16]. Bien que la correction chirurgicale des lésions gauches puisse normaliser la pression veineuse pulmonaire, cette vasoconstriction artérielle persiste dans le postopératoire, même si elle tend à diminuer avec le temps.
| Hypertension artérielle pulmonaire (HTAP) (I) |
|
Définition de l'HTAP: PAP moy > 25 mmHg au repos et RAP > 240 dynes•s•cm-5 (valeur normale : 60-150 dynes•s•cm-5).
Classification de l'OMS:
- HTAP essentielle: HTAP primaire, HTAP des cardiopathies congénitales, HTAP secondaire (médicaments, obésité, âge)
- HTP postcapillaire (insuffisance ventriculaire gauche, valvulopathie mitrale)
- HTAP due à l'hypoxie alvéolaire (BPCO, SAS, haute altitude)
- Maladie thrombo-embolique pulmonaire
- HTAP d’origine multifactorielle non éclaircie
Syndrome d'Eisenmenger: HTAP fixée aréactive, PAPm > 55 mmHg, RAP > 800 dynes•s•cm-5
L'hypertension pulmonaire postcapillaire est souvent associée à une vasoconstriction artériolaire secondaire (précapillaire).
|
Physiopathologie
En systole, l’arbre vasculaire pulmonaire doit absorber la totalité du volume systolique car la valve mitrale est fermée, raison pour laquelle les RAP sont dix fois plus basses que les RAS. La compliance artérielle est le fait de tout le circuit vasculaire pulmonaire, alors qu'elle n'est liée qu'à l'élasticité de l'aorte dans l'arbre systémique. Le nombre d'artérioles dans le circuit pulmonaire est 10 fois plus élevé que dans le circuit systémique; la compliance y est également 10 fois plus importante. Lorsque le débit cardiaque s'élève à l’effort, la compliance augmente de 30%, et les résistances vasculaires pulmonaires baissent afin de contenir cet excès de volume, mais la diminution des RAP ne peut pas être importante vu que le lit pulmonaire est déjà en vasodilatation active au repos. La PAP s’élève donc à l’effort intense, mais la PAPm reste normalement en-dessous de 30 mmHg. Une élévation > 3 mmHg par L/min de débit cardiaque signe une réponse anormale à l'exercice [34]. Une augmentation excessive de la PAP à l’exercice est un signe précoce de l’HTAP. Par contre, son augmentation au repos est un signe tardif puisqu’il faut que 50% de la circulation soit obstruée pour qu’elle reste élevée en permanence [33].
La postcharge du VD consiste en trois composantes : la résistance artériolaire fixe, l’impédance pulsatile et la compliance vasculaire. En clinique, on ne calcule que la première (RAP = (PAPm – PAPO)/DC), qui mesure la résistance moyenne comme si le flux était continu ; elle représente 50-70% de la résistance à l’éjection [31,60]. La deuxième quantifie la postcharge dynamique du VD puisqu’elle traduit l’opposition au flux dans un système pulsatile (Z = Pinst/flux) ; elle peut se calculer à partir du flux Doppler pulsé dans l’AP et de la mesure simultanée de la PAP. Elle représente le tiers de la postcharge du VD. Comme elle inclut la résistance et la rigidité des vaisseaux pulmonaires, elle offre une meilleure corrélation avec la survie que les RAP seules [39]. L'installation rapide d'une hypertension pulmonaire comme on le voit en haute altitude pénalise le VD qui baisse sa performance systolique d'environ 30% [2].
La compliance artérielle est le rapport entre le volume systolique et la pression pulsée (Ca = VS/PP, où PP = PAPs – PAPd) (voir Tableau 5.5). L’arbre vasculaire pulmonaire est un système à basse résistance et à haute compliance. Lorsque cette dernière diminue, la différentielle systolo-diastolique, ou pression pulsée, augmente. Comme la compliance et la résistance varient en sens inverse, leur produit (R • C, en 1/seconde) reste constant ; il représente la constante de temps qui caractérise la baisse de la PAP en diastole [31] ; il reste stable lorsque le traitement de l’HTAP est efficace, alors que l’évolution naturelle de la maladie entraîne une baisse de compliance plus importante que l’augmentation des RAP. La compliance est donc un marqueur plus fin du degré d’HTAP : un rapport VS/PP < 0.81 mL/mmHg prédit une probabilité de survie à 4 ans de < 40%, alors qu’un rapport > 2.0 prédit une survie de 100% [36]. D’ailleurs, la rigidité de l’arbre vasculaire (baisse de la compliance) est le principal déterminant lié à la diminution des indices de fonction du VD [55]. La relation qui unit la résistance et la compliance n’est toutefois pas linéaire, mais curvilinéaire et logarithmique [32]. Ainsi de petites variations de résistance se traduisent par de larges variations de la compliance lorsque les RAP sont basses et que la courbe est redressée, alors que les mêmes variations de résistance modifient peu la compliance lorsque les RAP sont élevées parce que la courbe est assez plate. Les vasodilatateurs pulmonaires sont donc plus efficaces au début de la maladie qu’en phase terminale.
Contrairement à l’image que l’on rencontre dans les artères systémiques (voir Figure 5.63), les courbes de pression et de flux sont pratiquement identiques dans l’artère pulmonaire, parce qu’il existe normalement peu d’onde de pression se réfléchissant en périphérie dans l’arbre vasculaire pulmonaire qui est vasodilaté en permanence (pression réfléchie : voir Couplage ventriculo-artériel). Lorsque les RAP augmentent, cette configuration se modifie : dans l’HTAP, la courbe de pression et celle de flux se dissocient et prennent la même allure qu’en voie systémique. Cela est dû à l’installation d’une onde de pression réfléchie par l’élévation des résistances artériolaires pulmonaires; cette onde de pression apparaît sur la courbe de PAP comme un pic de surpression dans la deuxième moitié de la systole; elle s’ajoute à la pression d’éjection du VD et augmente donc la postcharge réelle du ventricule [9]. La silhouette de la courbe de pression systolique en AP illustre ainsi le degré de rigidité des vaisseaux pulmonaires: le pic de pression systolique de la phase protosystolique se double d'un deuxième pic plus élevé pendant la phase télésystolique; celui-ci ajoute une valeur de 10 à 30 mmHg à la PAP (Figure 5.125) [51]. Son intensité est corrélée à la péjoration de l'état clinique des malades [29].
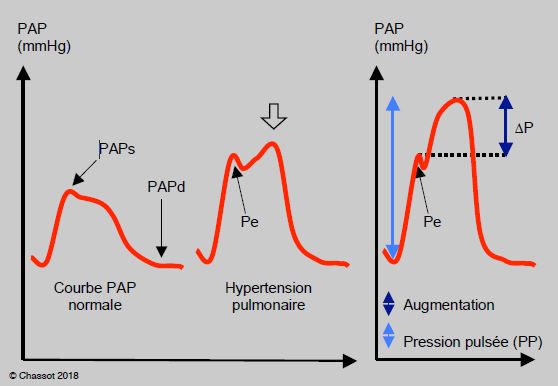
Figure 5.125 : Couplage entre le VD et la circulation pulmonaire. La réflexion de l’onde de pression due à la rigidité de l’arbre pulmonaire et à l’élévation des RAP donne lieu à une augmentation significative (ΔP) de la pression systolique par rapport à la pression avec laquelle le flux est éjecté du VD dans l’AP (Pe, pression d’entrée); cette augmentation de la pression télésystolique élève la postcharge du VD. L’index d’augmentation (ΔP/PP) s‘accroît avec la rigidité des vaisseaux. Le crochetage Pe correspond au “notching” typique de l’HTAP sur le flux Doppler dans l’AP (échocardiographie) [9]. A: courbe d'AP normale. B: augmentation de la PAP. C: HTAP sévère. PAPs: pression artérielle pulmonaire systolique. PAPd: pression artérielle pulmonaire diastolique.
Les vaisseaux pulmonaires sont moins innervés que le circuit systémique et ont une répartition différente des récepteurs sympathiques ; les récepteurs α y sont rares et les récepteurs β prédominent. La stimulation sympathique a un effet préférentiellement β vasodilatateur lorsque les RAP sont basses, mais un effet α vasoconstricteur lorsque les RAP sont déjà élevées [56]. Les vasoconstricteurs comme la nor-adrénaline ou la phényléphrine ont moins d’effet sur la circulation pulmonaire que sur la circulation systémique [49]. La vasopressine n’augmente pas la PAP car le rapport entre les récepteurs V1 (vasoconstriction) et V2 (vasodilatation) est en faveur des seconds dans le lit pulmonaire; elle a un effet vasodilatateur pulmonaire à faible dosage, mais un effet vasoconstricteur seulement à forte dose [11]. Les inhibiteurs de la phosphodiestérase-3 (IPDE3) comme la milrinone agissent sur les artérioles précapillaires et préalvéolaires qui contiennent l'enzyme PDE-3; ils y induisent une vasodilatation. Cependant, cet effet peut être atténué dans les artérioles remaniées des patients souffranat d'HTAP [57].
Les vaisseaux pulmonaires sont maintenus dans une vasodilatation active permanente, qui est la résultante d’un équilibre dynamique entre plusieurs éléments.
- NO• : vasodilatateur sécrété localement par l’endothélium en fonction de la pulsatilité locale ; la NO-synthase transforme l’arginine en citrulline qui est le donneur de NO•.
- Prostacycline I2E1 : vasodilatation. La prostacycline est produite à partir de l’acide arachidonique par la cyclo-oxygénase endothéliale ; elle stimule l’adénylate-synthase qui transforme l’ATP en cAMP.
- Endothéline E1 : vasoconstriction.
- Thromboxane A2, sérotonine et angiotensine II : vasoconstriction.
- Hypoxie alvéolaire : vasoconstriction locale si PaO2 < 60 mmHg.
- Hypercapnie et acidose (élévation de la [H+] locale) : vasoconstriction.
Dans l’hypertension artérielle pulmonaire, la production de NO• est freinée, et l’activité des phosphodiestérases-5, qui catabolisent le GMPc, est augmentée. Ceci conduit à une vasoconstriction parce que le GMPc est le messager intracelulaire du NO• ; normalement, il abaisse la [Ca2+]i dans les cellules musculaires lisses et provoque une vasodilatation. Les vasodilatateurs pulmonaires dimiuent également l’adhésivité plaquettaire, alors que les vasoconstricteurs l’augmentent ; de plus, les vasoconstricteurs favorisent la prolifération cellulaire endothéliale, myoblastique et fibroblastique. La noradrénaline étant métabolisée par l’endothélium pulmonaire, ses concentrations sont plus élevées en cas d’hypertension pulmonaire [30].
Le fonctionnement du VD est au centre de la physiopathologie clinique de l’HTAP. Face à l’augmentation chronique de sa postcharge, le VD dilate et s’hypertrophie (HVD) ; sa masse augmente jusqu’à 6 fois. La plasticité du VD est élevée, puisque l’hypertrophie débute quelques heures déjà après l’augmentation de postcharge [7]. Plus il est hypertrophié, plus le VD se comporte comme le VG. La proportion de raccourcissement circulaire augmente par rapport au raccourcissement longidutinal, qui devient minoritaire: la contraction de type péristaltique est perdue. Sa courbe de Starling se redresse et il devient tolérant à l’augmentation de postcharge, mais son débit devient dépendant de la précharge ; il ne peut plus amortir les variations du retour veineux en maintenant le débit pulmonaire constant. L’hypovolémie conduit à une baisse du débit pulmonaire et à une hypoxémie. En diastole, la pression du VD hypertrophié et surchargé est supérieure à celle du VG ; le septum interventriculaire bombe dans le VG et réduit le remplissage diastolique gauche (voir Figure 5.114) [28]. Malgré l'expression de gènes fœtaux facilitant son hypertrophie, le VD est moins performant que le VG comme pompe-pression: son pic de pression est atteint plus tardivement, d'où une désynchronisation des deux ventricules et une contraction postsystolique persistante à droite alors que le VG est déjà en diastole.
La circulation pulmonaire et le ventricule droit forment un tout fonctionnel qu’il est important d’étudier ensemble en fonction du couplage ventriculo-artériel [9]. Pour atteindre de hautes valeurs de PAP, il faut la conjonction de deux éléments : des résistances artériolaires élevées et une force propulsive suffisante. Celle-ci est fournie par le VD, dont la taille et l’hypertrophie ont une forte valeur pronostique pour la survie des patients. Ainsi, l’HTAP est fonction de la capacité du VD à générer chroniquement de hautes pressions pulmonaires. Une PAP de 80/40 mmHg signifie que le VD est capable de soutenir ce régime de pression en augmentant sa force contractile de 3 à 5 fois par hypertrophie [61]. Une augmentation de la PAP de 30 mmHg à l'effort (PAP de base 50 mmHg) traduit une réserve contractile de meilleur pronostic qu'une faible élévation de pression [20]. La défaillance droite se traduit au contraire par l’impossibilité de travailler contre une telle postcharge : la PAP tend à redescendre, alors que les RAP continuent à augmenter et que la situation hémodynamique empire (voir Figure 5.121) [23]. La gravité et le pronostic de la maladie tiennent donc davantage à la fonction ventriculaire droite qu’à la valeur de la PAP en elle-même. La découverte d’une HTAP dans le préopératoire doit commander immédiatement une échocardiographie pour évaluer la fonction droite, car c’est elle qui va déterminer le risque opératoire, aussi bien en chirurgie cardiaque qu’en chirurgie non-cardiaque.
En situation aiguë, un VD normal peut soutenir une PAPsyst de 40-60 mmHg pendant 1-2 heures avant de défaillir et de se dilater. En situation chronique et progressive, l’adaptation du VD à une augmentation de sa postcharge se fait par une hypertrophie concentrique et une transformation vers une forme plus arrondie qui le rendent davantage dépendant de la précharge et moins de la postcharge. Cette adaptation lui permet d'augmenter sa contractilité de 4 à 5 fois [61]. Mais le VD ne peut surmonter longtemps un excès de résistance à l’éjection et il commence à se dilater pour utiliser au maximum l'effet Starling, ce qui augmente encore son stress de paroi et sa consommation d’O2, alors que sa perfusion coronarienne est davantage compromise [6]. La dilatation l’amène en butée contre le péricarde. Comme l’espace intrapéricardique est limité, l’augmentation de volume du VD fait basculer le septum interventriculaire dans la cavité gauche en diastole et limite le remplissage diastolique de celle-ci. Ainsi une surcharge de volume aggrave non seulement l’insuffisance congestive droite mais encore limite le remplissage et le volume d’éjection du VG. L’interaction diastolique par le déplacement du septum est au moins aussi importante que la chute de l'éjection du VD dans la genèse du bas débit systémique [7]. Avec l’hypertrophie et l’élévation de postcharge, la durée de la contraction droite se prolonge et le VD se contracte encore lorsque le VG est déjà en diastole, ce qui produit une bascule systolique du septum vers la gauche et une dyskinésie septale qui gène l’éjection des deux ventricules [61]. L’augmentation de la postcharge gauche par un vasoconstricteur tend au contraire à repousser le septum dans sa position normale convexe dans le VD, donc à retrouver l’appui du VG à l’éjection droite. La dilatation du VD (> 85 mL/m2, VS/VtsVD > 1) et de l’OD (S > 15 cm2/m, où m = hauteur de l’individu en mètre), l’élévation de la PVC (> 15 mmHg) et la baisse de la fonction systolique droite (FE < 0.35) sont les principaux critères autorisant une évaluation pronostique (voir Insuffisance ventriculaire droite) [61]. L’insuffisance cardiaque droite se caractérise par un bas débit et par une élévation de la pression veineuse centrale au repos, avec rétention liquidienne, stase veineuse, oedèmes, ascite, hépatopathie et insuffisance rénale. L’élévation de la POD > 15 mmHg peut rouvrir un foramen ovale perméable et occasionner un shunt droite – gauche cyanogène. La mortalité de la défaillance droite dans le cadre de l’HTAP s’élève jusqu’à 40% [24].
| Hypertension artérielle pulmonaire (II) |
|
Facteurs étiopathogéniques dans la paroi de l'arbre vasculaire pulmonaire:
- Dysfonction endothéliale favorisant la vasoconstriction: ↓ production de NO•, ↓ GMPc, excès de phospholipase C qui conduisent à une ↑ [Ca2+]i
- Modifications congénitales du fonctionnement des canaux Kv qui règlent la [Ca2+] dans les cellules musculaires lisses
- Prolifération cellulaire excessive et métabolisme dirigé vers la glycolyse
Lésions histologiques:
- Hyperplasie endothéliale et hypertrophie de la média
- Fibrose intimale concentrique
- Microthromboses
- Lésions plexiformes (HTAP précapillaire)
Face à l'augmentation de sa postcharge, le VD s'hypertrophie. Sa fonction systolique est l'élément-clef pour générer une haute pression pulmonaire. Lorsqu'il défaille, la PAP baisse alors que les RAP continuent à augmenter. La fonction du VD mesure mieux le risque clinique que la valeur de la PAP.
|
Conséquences pour la prise en charge clinique
Les patients souffrant d’hypertension pulmonaire se caractérisent par une perte complète de compliance hémodynamique dans la circulation droite. Ils présentent une physiopathologie particulière [5,10,14].
- Le débit pulmonaire est abaissé et relativement fixe ; il ne peut pas augmenter proportionnellement à la demande en O2, d’où cyanose à l’effort. Toute élévation du débit cardiaque se traduit par une élévation importante de la PAP.
- Face à l’augmentation chronique de sa postcharge, le VD se dilate et s’hypertrophie (HVD). Plus il est hypertrophié, plus le VD se comporte comme le VG ; il tolère l’augmentation de postcharge mais son débit devient dépendant de la précharge. Il ne peut plus amortir les variations du retour veineux en maintenant le débit pulmonaire constant ; l’hypovolémie conduit à une baisse du débit pulmonaire et à une hypoxémie.
- En diastole, le bombement du septum interventriculaire dans le VG réduit le remplissage gauche et le volume systolique systémique ; l’élévation de la postcharge gauche (vasoconstriction artérielle systémique) tend à replacer le septum dans sa position physiologique.
- Une hypotension systémique peut compromettre la perfusion coronarienne droite en réduisant la composante systolique du flux coronaire vers le VD ; un vasoconstricteur systémique est requis pour parer au risque ischémique.
- En cas de foramen ovale perméable, un shunt droite → gauche cyanogène peut s’installer à la faveur d’une augmentation excessive de la POD.
- Malgré l’épaississement des parois artérielles pulmonaires, les petits vaisseaux artériolaires périphériques conservent une réactivité vasculaire ; les RAP peuvent encore augmenter par hypoxémie, hypercarbie, acidose, hypothermie ou stress sympathique [8].
Lors de la prise en charge de ces malades en salle d'opération ou aux soins intensifs, il est capital d'éviter toutes les situations qui peuvent augmenter les RAP:
- Hypoventilation (hypercarbie, hypoxémie, atélectasies);
- Surpression intrathoracique (variable selon la fonction du VD, respecter une hyperventilation normobarique);
- Acidose;
- Hypothermie;
- Stimulation sympathique (stress, douleur);
- Anémie aiguë (seuil de transfusion Hb ≥ 100 gm/L) ; la baisse du transport d’O2 ne peut être compensée que par une augmentation du débit pulmonaire, ce qui n'est pas possible car celle-ci élèverait considérablement la PAP et la fonction du VD ne permet pas de lutter contre une postcharge si haute.
Nosologie de l’HTAP
La symptomatologie de l’HTAP est peu spécifique : dyspnée, hypoxémie, cyanose d’effort, syncope, pseudo-angor, signes d’insuffisance ventriculaire droite. Elle peut s’accompagner d’hémoptysies. La polycythémie compense la baisse de la PaO2. L’ECG montre des signes d’HVD : déviation droite, grandes ondes R en V1 et V2, BBD. La radiographie du thorax décèle une dilatation des artères pulmonaires, du VD et de l’OD (coeur globulaire). L’échocardiographie montre une dilatation et une hypertrophie du VD, un aplatissement et un mouvement paradoxal du septum interventriculaire, et une insuffisance tricuspidienne. Les critères de sévérité de l’HTAP sont les suivants [10,41]:
- PAPm > 35 mmHg;
- SaO2 < 90%;
- Hb > 150 gm/L;
- Dysfonction du VD;
- NT-pro-BNP > 1'200 pg/mL.
Le degré de réversibilité de l’HTAP est évalué en préopératoire par un test au NO• (inhalation de 10-30 ppm au masque), à l’adénosine (6-12 mg iv) ou à l’époprosténol (> 2 ng/kg/min). On considère que l’HTAP est encore réversible si la PAPm baisse de > 25% (> 10 mmHg) et les RAP de > 30% (< 400 dynes•s•cm-5) sans modification du débit cardiaque [5].
La mortalité annuelle moyenne de l’HTAP est de 9% [4]. Bien que l’HTAP soit un facteur de risque majeur en chirurgie cardiaque et non-cardiaque (mortalité 5-25%, morbidité 40%), c’est surtout la fonction du VD qui est déterminante pour le pronostic : la mortalité est proportionnelle au degré de dysfonction du VD, et non directement liée à la valeur de la pression pulmonaire [47].
L’HTAP de haute altitude
Au sommet du Mont-Blanc (4'810 m) la PaO2 est de 75 mmHg. Jusqu'à 7'000 mètres, le contenu artériel en O2 est maintenu grâce à l'élévation de l'Hb (18-19 g/L), bien que la SaO2 diminue (88% à 5'000 m, 75% à 7'000 m) [17]. A l’Everest (8‘848 m), la PaO2 est de 25-36 mmHg, la SaO2 de 54% et la PaCO2 de 11-15 mmHg [17,37]. A ces valeurs, le poumon subit une vasoconstriction hypoxique massive (RAP augmentée de 100 à 300%). Normalement, la vasoconstriction pulmonaire hypoxique, qui est réglée par la pO2 alvéolaire, détourne le sang artériel des zones hypoventilées (atélectasies) vers les zones normoventilées ; elle diminue la désaturation artérielle liée à l’effet shunt (Figure 5.126). Cette vasoconstriction pulmonaire hypoxique est un phénomène rapide qui s’installe entre 20 secondes à 2 minutes [42,62]. En altitude, comme dans l’apnée du sommeil (SAS) et chez l’obèse morbide, elle se généralise à l’ensemble des poumons, mais reste inhomogène [3]. Très variable selon les individus, elle se déclenche dès 2'500 m. Elle est d’autant plus intense que l’ascension est rapide et les efforts importants.
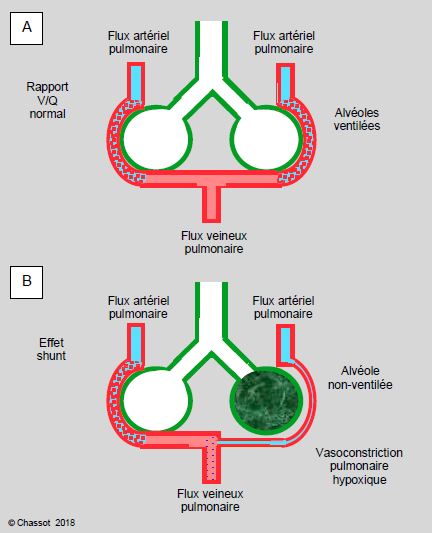
Figure 5.126 : Vasoconstriction pulmonaire hypoxique. A : ventilation et perfusion pulmonaires normales ; le rapport V/Q est normal. B : lorsque des alvéoles ne sont plus ventilées, les artérioles qui les vascularisent se vasoconstrictent pour freiner le débit sanguin et limiter l’effet shunt qui résulte de cet afflux de sang désaturé dans la veine pulmonaire.
Le phénomène se passe dans les celllules de la musculature lisse (Figure 5.127). Par leur chaîne d’oxydo-réduction, les mitochondries agissent comme des senseurs pour la pO2 alvéolaire. En effet, elles produisent normalement des Reactive Oxygen Species (ROS), qui sont des superoxydes sécrétés en petite quantité. Ces ROS activent des canaux membranaires voltage-dépendants extrayant le potassium de la cellule et la maintenant hyperpolarisée (canaux KV) ; cette hyperpolarisation physiologique inhibe les canaux calciques voltage-dépendants (canaux Ca2+ L), donc moins de Ca2+ entre dans la cellule ; la [Ca2+]i reste basse, donc la contraction musculaire est freinée. Physiologiquement, les vaisseaux pulmonaires sont maintenus en vasodilatation active par hyperpolarisation membranaire. Mais si la pO2 enregistrée par les mitochondries est trop basse à cause de l’hypoxie, la production de ROS est faible et les canaux KV ne sont plus activés : la membrane est moins hyperpolarisée, l’activité des canaux Ca2+ L augmente et la [Ca2+]i s’élève ; la troponine C est stimulée et la musculature se contracte [40,42].
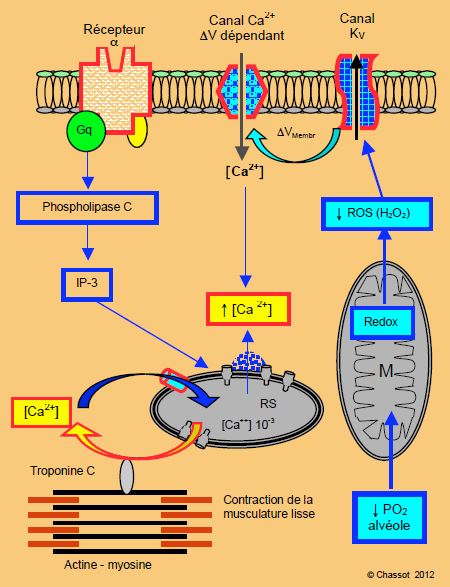
Figure 5.127 : Mécanismes cellulaires mis en jeu dans la vasoconstriction pulmonaire hypoxique liée à la haute altitude. L’hypoxie freine la chaîne d’oxydo-réduction mitochondriale (Redox), ce qui diminue la production de superoxydes (ROS : Reactive Oxygen Species) ; ceux-ci n’activent plus suffisamment les canaux potassiques Kv qui maintiennent la membrane hyperpolarisée. De ce fait, le Ca2+ entre en plus grande quantité par les canaux calciques L et accroît l’activité de la musculature lisse vasoconstrictrice.
Le deuxième phénomène en jeu est la production de NO• par l’endothélium vasculaire des poumons. Le NO• est un puissant vasodilatateur, dont la production est réglée par le flux pulmonaire : elle augmente lorsque celui-ci est élevé pour permettre un haut débit sans variations de pression. Dans les zones hypoxiques, sa production est abaissée puisqu’il fait partie de l’ensemble des ROS, dérivés superoxides produits au cours de l’oxydo-réduction de l’O2.
Chez les alpinistes séjournant en haute altitude, l’HTAP d’altitude évolue en deux phases [19,52].
- Phase I : la PAPsyst s’élève jusqu’à 50-80 mmHg ; la PAP moyenne est de 20 mmHg (5'000 m) à 35 mmHg (7'000 m) au repos, et respectivement de 40 et 55 mmHg à l’effort. La vasoconstriction pulmonaire hypoxique est intense, la production de NO• est abaissée et celle d’endothéline E1 est augmentée. Comme la vasoconstriction est inhomogène, l’oedème alvéolaire s’installe dans les zones non-vasoconstrictées. L’OAP d’altitude est lié à deux phénomènes :
- PAP excessive dans les zones non vasoconstrictées;
- Défaut de transport du Na+ et de l’eau dans l’épithélium alvéolaire rendant inefficace le système qui réabsorbe activement l’oedème alvéolaire.La faible production de NO• et le défaut de clairance du liquide alvéolaire sont des éléments constitutionnels génétiques. L’augmentation de l’hématocrite avec l’acclimatation permet un transport d’O2 adéquat, et de bonnes performances physiques sont encore possibles.
- Phase II : après 4-6 semaines d’acclimatation pendant lesquelles l’HTAP s’est homogénéisée et a protégé l’alpiniste de l’OAP, le VD entre dans une phase d’insuffisance ventriculaire dû à l’excès de postcharge de longue durée. Cette phase est souvent accompagnée de broncho-pneumonie.
L’œdème pulmonaire d’altitude (High altitude pulmonary edema, ou HAPE) survient en général pendant les premiers jours d’acclimatation. Les lésions pulmonaires sont inhomogènes : certaines zones sont en vasoconstriction hypoxique intense, alors que d’autres ne sont pas vasoconstrictées. De ce fait, ces dernières sont perfusées sous haute pression, et ne sont pas protégées par la vasoconstriction [3]. La pression capillaire y est ≥ 25 mmHg et le liquide interstitiel fuit dans les alvéoles : c’est l'OAP. A ce mécanisme s’ajoutent des efforts d’hyperinflation pulmonaire et d’hyperventilation compensatoire qui créent une dépression intrathoracique profonde en inspirium ; l’afflux de sang dans les poumons est massif, comme dans une manœuvre de Mueller. L'augmentation de postcharge induit une dysfonction du VD, qui perd 30% de sa capacité fonctionnelle en 2 semaines à 4'500 mètres [2]. Il existe une grande variabilité interindividuelle dans l’incidence de l’OAP d’altitude ; seul un petit nombre d’individus y est susceptible. Ce sont ceux qui présentent les plus hautes pressions pulmonaires en réaction à l’hypoxie. Il s’agit probablement de personnes génétiquement prédisposées à une moindre production de NO• et à un défaut d'évacuation liquidienne alvéolaire [3,52]. Il existe aussi des variations intra-individuelles : certains alpinistes ont souffert une fois d’OAP alors qu’ils avaient déjà accompli plusieurs ascensions à 8'000 mètres sans problèmes.
Les autochtones qui vivent en altitude dans les Andes ou l'Himalaya (3’500-4'500 m) ont différents mécanismes adaptatifs [18,35,48].
- Diminution de la vasoconstriction pulmonaire hypoxique et augmentation de la production pulmonaire de NO• ; les Tibétains vivant à 4'000 m n’ont pratiquement pas d’HTAP.
- Remodelage de l’activité des canaux KV et Ca2+ L.
- Normalisation de la [Ca2+]i dans les cellules musculaires lisses à une pO2 intracellulaire basse.
- Muscularisation réversible des artérioles pulmonaires (hypertrophie) et épaississement de l’endothélium (hyperplasie); les RAP augmentent discrètement, mais le risque d’OAP diminue. Ce phénomène est plus marqué chez les mammifères (yacks, chèvres) que chez les humains.
- Hypertrophie ventriculaire droite.
Outre le traitement de l’OAP, la thérapeutique consiste à augmenter la PaO2 (oxygène) et la pression (caisson hyperbare, redescente de 500 à 1000 m). Les anti-phosphodiestérases-5 (sildenafil, tadalafil) et les anti-endothéline-1 (bosentan) diminuent l’HTAP d’altitude de 25-30% ; les agents β-adrénergiques améliorent la réabsorption du liquide alvéolaire [50,52]. La dexaméthasone agit comme anti-inflammatoire et anti-œdémateux. Les anticalciques (nifédipine) peuvent être utiles chez les répondeurs, mais ils ne sont que 20% de la population.
L’HTAP des congénitaux
Sa cause la plus fréquente est un shunt gauche - droit non restrictif. Elle survient dans 50% des cas de communication interventriculaire (CIV) et de canal atrio-ventriculaire, où il y a surcharge de volume et de pression, mais dans seulement 10% des communications interauriculaires (CIA), où il y a surcharge de volume uniquement [10]. Elle apparaît dans l'enfance déjà lors de CIV, mais seulement à l'âge adulte en cas de CIA [43]. Le stress pariétal vasculaire provoqué par le flux pulmonaire excessif cause une extension de la musculature lisse dans des vaisseaux périphériques qui ne sont normalement pas musculaires; à ce stade, la pression reste normale. L'hypertension progressive est associée à une hypertrophie de la média des artères plus proximales, une restriction de la lumière et une artérite nécrosante avec des lésions plexiformes [46]. Finalement, le nombre des vaisseaux distaux diminue et la fibrose est massive ; l'HTAP est fixée et irréversible. C'est le syndrome d'Eisenmenger, caractérisé par une PAPmoy > 50 mmHg et des résistances vasculaires pulmonaires > 800 dynes•cm•s-5. Le flux à travers le shunt gauche - droit est alors bidirectionnel ou renversé; c'est la principale origine de l'apparition d'une cyanose chez les congénitaux adultes [12]. Une baisse de pression systémique par hypovolémie ou par vasodilatation artérielle aggrave la composante droite - gauche du shunt, donc la cyanose. La correction chirurgicale n'est plus possible lorsque la rapport RAP / RAS est supérieur à 0.7. En présence d'HTAP, la survie des patients souffrant de cardiopathie congénitale est meilleure que celle des patients souffrant d'HTAP primaire de l’adulte: elle est de 80% à 10 ans pour les patients souffrant d’Eisenmenger, mais de 30% à 5 ans pour ceux souffrant de maladie thrombo-embolique [23,26]. La raison en est probablement le maintien des caractéristiques fétales du myocarde droit, qui lui permettent d’assumer une postcharge élevée sur le long terme (voir Chapitre 15 Hypertension pulmonaire).
Ventilation et hypertension pulmonaire
Pour l’anesthésiste et l’intensiviste, ventiler les patients souffrant d’HTP est souvent un défi clinique. La ventilation mécanique de ces malades est un compromis entre une hyperventilation active pour baisser les RAP et le maintien d'une pression intrathoracique moyenne (Pit) basse pour éviter une augmentation de postcharge droite. Si le volume courant est faible, on risque des atélectasies, une hypercarbie, et une augmentation des RAP dans les petits vaisseaux péri-alvéolaires; s'il est élevé, l'hyperinflation augmente la Pit et comprime les gros vaisseaux extra-alvéolaires par la distension des alvéoles pulmonaires [14,27]. Le volume courant idéal correspond à celui de la capacité résiduelle fonctionnelle (CRF) (voir Figure 5.101). Il faut jouer sur le volume courant, la fréquence et le mode ventilatoire pour obtenir la PaCO2 et la Pit moy les plus basses possible. De ce point de vue, la durée de l'inspirium augmente davantage la Pit moy que la valeur du pic inspiratoire de pression.
Toutefois, deux phénomènes importants viennent faciliter la tâche du clinicien.
- La compression des gros vaisseaux extra-alvéolaires, dans la partie droite du graphique de la Figure 5.101, ne s’applique pas réellement au patient souffrant d’HTAP, parce que la paroi épaissie et rigide de ses vaisseaux pulmonaires empêche toute compression par une ventilation à haut volume courant. Il n’y a donc pas lieu de craindre une augmentation significative de la PAP lors d’une hyperventilation mécanique.
- L'accroissement de postcharge pour le VD que représente l'IPPV est très faible par rapport à sa postcharge habituelle: ajouter une Pit moy de 10 mmHg à une PAPmoy de 50 mmHg modifie moins les conditions hémodynamiques que lorsque la PAPmoy est normale (20 mmHg). Le risque de décompensation du VD est donc très faible lorsque les pressions ventilatoires restent dans les limites habituelles [10].
De plus, l'IPPV offre la possibilité d'hyperventiler le patient et, ce faisant, de diminuer ses RAP par alcalose respiratoire. Seule une dysfonction droite isolée sans élévation chronique de la pression pulmonaire présente un risque de décompensation majeure lors d’IPPV, mais non la situation d’une HTP chronique accompagnée d’une hypertrophie ventriculaire droite [14]. La réaction hémodynamique du patient à l'IPPV peut être testée en préopératoire en lui faisant réaliser une manoeuvre de Valsalva une fois le cathéter artériel en place et en observant l'évolution de la pression artérielle. Le plus souvent, les variations respiratoires sont atténuées et la pression moyenne (PAM) est stabilisée à une valeur très légèrement inférieure (< 15%) à sa valeur en spontanée. Ceci laisse présager une bonne tolérance à l’IPPV.
| Hypertension artérielle pulmonaire (III) |
|
Le pronostic de l'HTAP sévère (mortalité annuelle 15%) tient davantage à la fonction ventriculaire droite qu'à la valeur de la pression pulmonaire.
Critères de risque périopératoire en cas d'HTAP:
- PAPmoy > 35 mmHg
- SaO2 < 90%
- Hb > 150 gm/L
- Dysfonction ventriculaire droite
Facteurs aggravants: hypoxie, hypercarbie, atélectasie, acidose, hypothermie, stress, douleur.
Impact clinique de l'HTAP:
- Débit pulmonaire fixe, hypoxémie à l'effort
- Maintien de la réactivité des petits vaisseaux (vasoconstriction pulmonaire hypoxique)
- HVD: débit droit dépendant de la précharge, intolérance à l'hypovolémie
- Risque ischémique du VD élevé en cas d'hypotension systémique
- Insuffisance diastolique du VG (effet Bernheim)
- Shunt D → G si foramen ovale perméable
Les RAP augmentent en cas d'hypoventilation (vasoconstriction pulmonaire hypoxique) et en cas de ventilation à haut volume courant (VC) (distension et occlusion des vaisseaux par la distension alvéolaire). Mais en cas d'HTAP avec HVD, trois éléments favorisent l'IPPV:
- La paroi épaisse et rigide des vaisseaux pulmonaires empêche leur compression à haut VC
- L'IPPV représente un faible accroissement de postcharge pour le VD en cas d'HVD
- L'hyperventilation permet une certaine vasodilatation artériolaire pulmonaire
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2019
Références
- ARCHER SL, WEIR K, WILKINS MR. Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation 2010; 121:2045-66
- BARTELS K, BROWN D, FOX DL, et al. Right ventricular longitudinal strain is depressed in a bovine model of pulmonary hypertension. Anesth Analg 2016; 122:1280-6
- BÄRTSCH P, MAIRBÄURL, MAGGIORINI M, et al. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol 2005; 98:1101-10
- BENZA RL, MILLER DP, GOMBERG-MAITLAND M, et al. Predicting survival in pulmonary arterial hypertension. Insights from the Registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL). Circulation 2010; 122:164-72
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Anesthesiology 2003; 99:1415-32
- BOGAARD HJ, ABE K, VONK NOORDEGRAAF A, et al. The right ventricle under pressure. Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009; 135:794-804
- BRONICKI RA, BADEN HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 2010; 11(Suppl):S15-S22
- BUNDTS W, VAN PELT N, GILLYNS H, et al. Residual pulmonary vasoreactivity to inhaled nitric oxide in patients with severe obstructive pulmonary hypertension and Eisenmenger syndrome. Heart 2001; 86:553-9
- CHAMPION HC, MICHELAKIS ED, HASSOUN PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit. Circulation 2009; 120:992-1007
- CHASSOT PG, BETTEX DA. Anesthesia and adult congenital heart disease. J Cardiothorac Vasc Anesth 2006; 20: 414-37
- CURRIGAN DA, HUGHES RJ, WRIGHT CE, et al. Vasoconstrictor responses to vasopressor agents in human pulmonary and radial arteries: an in vitro study. Anesthesiology 2014; 121:930-6
- DALIENTO L, SOMERVILLE J, PRESBITERO P, et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J 1998; 19:1845-55
- FANG JC, DE MARCO T, GIVERTZ MM, et al. World Health Organization Pulmonary Hypertension Group 2: Pulmonary hypertension due to left heart disease in the adult – a summary statement from the Pulmonary Hypertension Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2012; 31:913-33
- FISCHER LG, VAN AH, BURKLE H. Management of pulmonary hypertension: physiological and pharmacological considerations for anesthesiologists. Anesth Analg 2003; 96:1603-16
- GALIÉ N, HUMBERT M, VACHIERY JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the diagnosis and treatment of pulmonary hypertension of the ESC and the ERS, endorsed by the AEPC and ISHLT. Eur Heart J 2016; 37:67-119
- GERGES C, GERGES M, LANG MB, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in "out-of-proportion" pulmonary hypertension. Chest 2013; 143:758-66
- GROCOTT MPW, MARTIN DS, LEVETT DZH, et al. Arterial blood gases and oxygen content in climbers on Mount Everest. N Engl J Med 2009; 360:140-9
- GROVES BMT, DROMA T, SUTTON JR, et al. Minimal hypoxic pulmonary hypertension in normal tibetans at 3’658 m. J Appl Physiol 1993; 74: 312-8
- GROVES BMT, REEVES JT, SUTTON JR, et al. Operation Everest II: elevated high altitude pulmonaryresistence unresponsive to oxygen. J Appl Physiol 1987; 63: 521-30
- GRÜNIG E, TIEDE H, ENYIMAYEV EO, et al. Assessment and prognostic relevance of right ventricular contractile reserve in patients with severe pulmonary hypertension. Circulation 2013; 128:2005-15
- GUAZZI M, BORLAUG BA. Pulmonary hypertension due to left heart disease. Circulation 2012; 126:975-90
- GUAZZI M, NAEIJE R. Pulmonary hypertension in heart failure. Pathophysiology, pathobiology, and emerging clinical perspectives. J Am Coll Cardiol 2017; 69:1718-34
- HADDAD F, DOYLE R, MURPHY DJ. HUNT SA. Right ventricular function in cardiovascular disease, Part II. Pathophysiology, clinical importance and management of right ventricular failure. Circulation 2008; 117:1717-31
- HADDAD F, KUDELKO K, MERCIER O, et al. Pulmonary hypertension associated with left heart disease: Characteristics, emerging concepts, and treatment strategies. Progr Cardiovasc Dis 2011; 54:154-67
- HOEPER MM, BOGAARD HJ, CONDLIFFE R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D42-50
- HOPKINS WE, OCHOA LL, RICHARDSON GW, TRULOCK EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996; 15:100-5
- HUGHES JMB, GLAZIER JB, MALONEY JE, et al. Effect of lung volume on the distribution of pulmonary blood flow in man. Resp Physiol 1968; 4:58-67
- KERBAUL F, RONDELET B, COLLART F, et al. Hypertension artérielle pulmonaire en anesthésie-réanimation. Ann Fr Anesth Réa 2005; 24:528-40
- KJELLSTRÖM B, MANOURAS A, WIKSTRÖM G. Right ventricular wave reflection relate to clinical measures in pulmonary arterial hypertension. Scand Cardiovasc J 2015; 49:235-9
- KWAK YL, LEE CS, PARK YH, et al. The effect of phenylephrine and norepinephrine in patients with chronic pulmonary hypertension. Anaesthesia 2002; 57:9-14
- LANKHAAR JW, WESTERHOF N, FAES TJC, et al. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol 2006; 291:H1731-H1737
- LANKHAAR JW, WESTERHOF N, FAES TJC, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008; 29:1688-95
- LAU EMT, MANES A, CELERMAJER DS, GALIE N. Early detection of pulmonary vascular disease in pulmonary arterial hypertension: time to move forward. Eur Heart J 2011; 32:2489-98
- LEWIS GD, BOSSONE E, NAEIJE R, et al. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation 2013; 128:1470-9
- MAGGIORINI M. Cardiopulmonary interactions at high altitude. Pulmonary hypertension as a common denominator. In: ROACH RC, ed. Hypoxia:through the lifecycle. New York: Kluwer Academic / Plenum Publisher, 2003
- MAHAPATRA S, NISHIMURA RA, SORAJJA P, et al. Relationship of pulmonary arterial capacitance and mortality in idiopathic pulmonary arterial hypertension. J Am Coll Cardiol 2006; 47:799-803
- MALCONIAN MK, ROCK PB, REEVES JT, et al. Operation Everest II: gaz tensions in expired air and arterial blood at extreme altitude. Aviat Space Environ Med 1993; 64:37-42
- McLAUGHLIN W, ARCHER SL, BADESCH DB et al. ACC/AHA 2009 expert consensus document on pulmonary hypertension : A report of the American College of Cardiology Foundation Task Force on expert consensus documents and the American Heart Association. Circulation 2009; 119:2250-94
- McMURRAY JJ, ADAMOPOULOS S, ANKER SD, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart Failure. Eur Heart J 2012; 14:803-69
- MAUBAN JRH, REMILLARD CV, YUAN JXJ. Hypoxic pulmonary vasoconstriction: role of ion channels. J Appl Physiol 2005; 98:415-20
- MAURITZ GJ, RIZOPOULOS D, GROEPENHOFF H, et al. Usefulness of serial N-terminal pro-B-type natriuretic peptide measurements for determining prognosis in patients with pulmonary arterial hypertension. Am J Cardiol 2011; 108:1645-50
- MOUDGIL R, MICHELAKIS ED, ARCHER SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol 2005; 98: 390-403
- OECHSLIN E. Eisenmenger's syndrome. In: Gatzoulis MA, Webb GD, Daubeney PEF, Eds. Diagnosis and management of adult congenital heart disease. Edinburgh, Churchill Livingstone 2003, pp 363-77
- OPOTOWSKY AR. Clinical evaluation and management of pulmonary hypertension in the adult with congenital heart disease. Circulation 2015; 131:200-10
- PALEVSKY HI, SCHLOO BL, PIETRA CC, et al. Primary pulmonary hypertension: Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation 1989; 80:1207-21
- RABINOVITCH M, HAWORTH SG, CASTANEDA AR, et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 1978; 58:1007-22
- RAMAKRISHNA G, SPRUNG J, RAVI BS, et al. Impact of pulmonary hypertension on the outcomes of noncardiac surgery: predictors of perioperative morbidity and mortality. J Am Coll Cardiol 2005; 45:1691-9
- REMILLARD CV, YUAN JX. High altitude pulmonary hypertension: role of K” and Ca2+ channels. High Alt Biol Med 2005; 6:133-46
- RICH S, McLAUGHLIN VV. Pulmonary hypertension. In: ZIPES DP, et al, eds. Braunwald’s heart disease. A textbook of cardiovascular medicine. Philadelphia: Elsevier Saunders, 2005, 1807-42
- RICHALET JP, GRATADOUR P, ROBACH P, et al. Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension. Am J Resp Crit Care Med 2004; 171:275-81
- RUOHONIEMI DM, SISTA AK, DOANY CF, HEERDT PM. Perioperative pulmonary thromboembolism: current concepts and treatment options. Curr Opin Anaesthesiol 2018; 31:75-82
- SARTORI C, ALLEMANN Y, SCHERRER U. Pathogenesis of pulmonary edema: Learning from high-altitude pulmonary edema. Resp Physiol Neurobiol 2007; 159:338-49
- SIMONEAU G, ROBBINS IM, BEGHETTI M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009; 54:S43-S54
- SIMONEAU G, GATZOULIS MA, ADAIA I,, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62:S34-D41
- STEVENS GR, GARCIA-ALVAREZ A, SAHNI S, et al. RV dysfunction in pulmonary hypertension is independently related to pulmonary artery stiffness. JACC Cardiovasc Inaging 2012; 5: 378-87
- SUBRAMANIAM K, YARED JP. Management of pulmonary hypertension in the operating room. Semin Cardiothor Vasc Anesth 2007; 11:119-36
- TABUCHI A, STYP-REKOWSKA B, SLUTSKY AS, et al. Precapillary oxygenation contributes relevantly to gas exchange in the intact lung. Am J Respir Crit Care Med 2013; 188:478-81
- TÜLLER C, NICOD LP. Hypertension pulmonaire chronique. Forum Méd Suisse 2008; 8:812-7
- VACHIÉRY JL, ADIR Y, BARBERÀ JA, et al. Pulmonary hypertension due to left heart disease. J Am Coll Cardiol 2013; 62:D100-8
- VONK-NOORDEGRAAF A, HADDAD F, CHIN KM, et al. Right heart adaptation to pulmonary arterial hypertension. J Am Coll Cardiol 2013; 62:D22-33
- VONK NOORDEGRAAF A, WESTERHOF BE, WESTERHOF N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69:236-43
- WAYPA GB, SCHUMACKER PT. Hypoxic pulonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol 2005; 98:404-14