Anatomie et physiologie
Au repos, le débit coronaire est de 220 à 250 mL/min (4-5% du DC). Le flux est maintenu constant pour des PAM de 60 à 160 mmHg (régulation en 10-30 secondes). Il cesse dans les couches sous-endocardiques lorsque la PAM est < 50 mmHg [18]. Avec un poids de 250-280 gm (0.6% du poids du corps), le coeur reçoit en moyenne 75 mL/100gm/min de sang. L'arbre vasculaire coronarien comporte trois types de vaisseaux [10] :
- Vaisseaux proximaux épicardiques, dits de capacitance, sous régulation endothéliale et neuro-humorale; ils sont le plus touchés par l'athérosclérose; ils participent pour moins de 10% à la résistance vasculaire coronarienne; leur volume augmente de 25% pendant la systole.
- Vaisseaux pré-artériolaires de résistance (diamètre 100-500 μm); ils sont le principal élément de contrôle neurogène et humoral du flux coronaire et représentent 25-35% de la résistance vasculaire totale.
- Vaisseaux distaux artériolaires intramyocardiques (précapillaires de diamètre 20–100 μm), où intervient plus de 50% de la perte de charge et qui sont sous régulation métabolique.
Les deux premières catégories sont innervées par le système nerveux sympathique et parasympathique. Le flux est si bien apparié à la demande en O2 dans les vaisseaux résistifs distaux que l'extraction d'O2 peut rester constante à 70% sur une vaste plage de pression de perfusion et de demande métabolique différentes. Comme les vaisseaux sont maintenus vasoconstrictés au repos, leur vasodilatation soudaine permet une augmentation rapide du flux sanguin. Lors d'un exercice important, le flux peut tripler.
Pendant la systole, la puissante musculature du VG écrase le lit coronaire et y interrompt le flux; ce phénomène, qui représente 10-25% des résistances vasculaires totales des coronaires, est d'autant plus significatif que le réseau est plus dilaté [23]. La compression par la pression intraventriculaire est surtout marquée sur le plexus sous-endocardique et sur les vaisseaux pénétrants transmuraux verticaux, alors que le flux dans les vaisseaux parallèles aux fibres musculaires est momentanément accéléré par le "massage" musculaire (Figure 5.131). Les vaisseaux sous-épicardiques ne sont pas comprimés pendant la systole. Ainsi, le flux dans les troncs coronaires est biphasique au cours du cycle cardiaque: il présente un pic systolique et un pic diastolique. En systole, le flux représente moins de 20% du flux coronaire total dans les territoires gauches, alors qu'il compte pour 40 à 50% du flux total dans le VD (voir Figure 5.116). La compression musculaire, beaucoup plus faible dans la paroi du VD, est la principale cause de cet aspect du flux coronaire. En diastole, la décompression des vaisseaux intramyocardiques très compliants par la relaxation de la paroi ventriculaire crée une vague de succion qui aspire le sang en périphérie [12]. Cet élément est le principal accélérateur du flux coronarien en protodiastole; il est renforcé en cas d'hypertrophie ventriculaire comme dans la sténose aortique, parce que la compression est plus forte que dans une situation normale [24]. Dans les zones akinétiques, c'est exclusivement la pression intraventriculaire qui est responsable de la compression systolique [13]. La vélocité maximale du flux coronarien survient en protodiastole; elle est diminuée lors de dysfonction diastolique avec défaut de relaxation, comme c'est le cas lors d'ischémie ou d'hypertrophie ventriculaire.
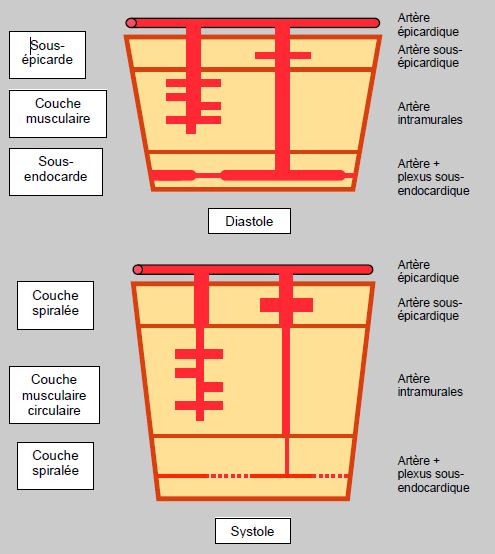
Figure 5.131 : Flux coronaire myocardique en diastole et en systole à travers les trois couches de la paroi ventriculaire. En systole, les flux épicardique et sous-épicardique sont maintenus, voire augmentés par reflux du sang comprimé en profondeur, alors que celui des artères intramurales, allongées et écrasées par la contraction de la musculature circulaire, diminue presque complètement. Dans le sous-endocarde, région adossée à la masse sanguine intracavitaire, la plus comprimée et la plus épaissie en systole, le flux est minime, voire nul en systole.
Alors que le flux est toujours antérograde pendant la diastole, on observe fréquemment une inversion du flux dans les vaisseaux intramyocardiques et épicardiques pendant la systole; le sang est de ce fait transloqué depuis le sous-endocarde vers les vaisseaux épicardiques qui agissent comme réservoir. Normalement, le rapport du flux sous-endocardique au flux épicardique est de 1.25:1 pendant la diastole. Lorsque le flux coronarien total est réduit de moitié par vasoconstriction, ce rapport tombe à 0.4 [15]. Le même phénomène survient lorsque le stress de paroi augmente (hypertension artérielle) ou en cas d'hypertrophie ventriculaire concentrique (sténose aortique). Ainsi la région sous-endocardique est la plus fragile, raison pour laquelle les mouvements électrocardiographiques de sous-décalage du segment ST sont si caractéristiques de l'ischémie. La région sous-endocardique est la zone la plus sensible à l’ischémie pour plusieurs raisons.
- Sa perfusion est exclusivement diastolique;
- Sa course de contraction radiaire et son degré de raccourcissement sont plus importants que ceux des couches plus externes, ce qui augment sa mVO2;
- En cas d’augmentation de postcharge, elle est la région la plus comprimée en systole;
- En cas d’augmentation de la précharge (Ptd élevée), elle est la région la plus comprimée en diastole, ce qui réduit son DO2;
- En cas d’hypertrophie myocardique, la distance entre les vaisseaux épicardiques et le sous-endocarde s’allonge et le DO2 distal diminue, alors que la mVO2 des myocytes hypertrophiés augmente.
La pression de perfusion coronaire (PPC) est définie comme la différence entre la pression diastolique aortique moyenne et la pression du sinus coronaire ou de la cavité ventriculaire gauche en télédiastole. La mesure de la pression aortique n'étant pas accessible en clinique, on recourt à la mesure de la pression artérielle moyenne (PAM) qui est la valeur la plus voisine de celle de la racine aortique [30,32] ; la pression télédiastolique du VG est remplacée par celle de l'OG évaluée par la PAPO:
PPC = PAM – PAPO
La PPC physiologique est > 50 mmHg ; en dessous de cette valeur, l’ischémie survient. La PAM doit donc rester > 65 mmHg pour assurer une perfusion myocardique globalement satisfaisante. Comme sa pression systolique est beaucoup plus basse que celle de l’aorte, le VD jouit d’une perfusion coronarienne systolo-diastolique ; 40-50% de son flux coronarien total est systolique.
Trois phénomènes règlent le débit coronarien [13] :
- L’autorégulation métabolique;
- Le contrôle neuro-humoral autonome (sympathique, parasympathique, catécholamines);
- L'endothélium vasculaire : action vasodilatatrice (NO•) ou vasoconstrictrice (endothéline).
L'autorégulation métabolique
Bien que la pression de perfusion puisse subir de grandes variations (PAM de 40 à 130 mmHg), l'autorégulation maintient un flux constant par un changement de résistance dans les artérioles, qui est proportionnel aux modifications de pression (Figure 5.132). La régulation a lieu instantanément, et revient à l'équilibre en 10-30 secondes [13]. Elle est plus importante dans les vaisseaux épicardiques que dans la zone sous-endocardique, ce qui contribue à la fragilité de cette dernière. Le flux est ainsi maintenu constant pour des pressions aortiques allant de 60 à 160 mmHg [6]. Le flux cesse dans les couches profondes lorsque la pression est inférieure à 50 mmHg (Pfz ou pression à flux zéro) [18]. Les courbes d'autorégulation du VG et du VD sont analogues. L'autorégulation est sous contrôle métabolique par plusieurs mécanismes (Tableau 5.10).
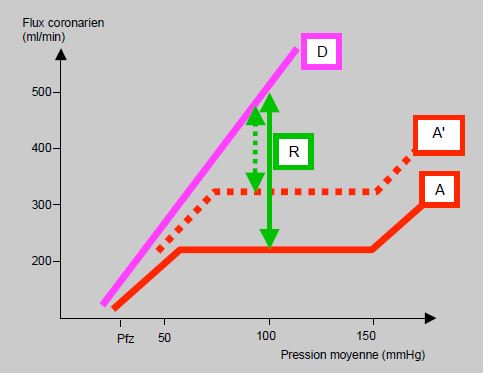
Figure 5.132 : Variation du flux coronaire en fonction de la pression de perfusion. A: courbe d'autorégulation; le flux est stable entre 60 et 160 mmHg de pression aortique. D: courbe à vasodilatation maximale; le flux est pression-dépendant. R: réserve coronarienne; c'est la différence entre le flux autorégulé et le flux à vasodilatation maximale pour une pression aortique donnée; chez un individu sain, la réserve coronarienne représente un flux triple par rapport au flux de repos. A': courbe d'autorégulation déplacée vers le haut lors d'anémie, d'hypertrophie ventriculaire ou d'effet inotrope positif; dans ce cas, la réserve coronarienne (en traitillé) est diminuée. Pfz: pression à flux zéro [18].
- L'adénosine joue un rôle vasodilatateur majeur; elle est libérée par les cellules lorsque la PO2 diminue en dessous des besoins métaboliques (hypoxie, ischémie, exercice) [22]; elle provient de la consommation des phosphates à haute énergie (ATP). Elle est inactivée par une désaminase; cette dernière est inhibée par le dipyridamole, qui provoque ainsi une vasodilatation massive par accumulation d'adénosine locale.
- Le NO• est sécrété par l'endothélium sain lorsque les forces de cisaillement augmentent (augmentation de pression et/ou flux). Si l'endothélium est endommagé, par exemple par l'athéromatose, la production de NO• est diminuée alors que la sécrétion d'endothéline vasoconstrictrice est augmentée.
- L'activation des canaux potassiques sous dépendance de l'ATP (canaux KATP) dans les cellules musculaires vasculaires hyperpolarise ces dernières et provoque une vasodilatation en faisant frein à l’entrée de Ca2+ dans le sarcoplasme; ces canaux sont normalement inhibés par l'ATP, et activés par l'adénosine [16].
- La PO2 et la PCO2 locales ont une influence au sein de certaines limites [6]; l'hypoxie et l'hypercarbie provoquent une dilatation; l'hyperoxie et l'hypocapnie entraînent une vasoconstriction [4]. L'hypocapnie sévère conduit à une diminution du flux coronaire plus importante que la diminution du débit cardiaque, ce qui occasionne un déséquilibre dans la balance DO2/VO2 myocardique.
- L'augmentation de la concentration locale de lactate et de [H+] induit une vasodilatation.
- L'ischémie aiguë, avec son cortège d'effets métaboliques, est un puissant stimulant vasodilatateur; elle provoque la libération d'adénosine et de NO•, et ouvre les canaux potassiques (KATP).
- Lors d’une augmentation de la demande en O2 ou lors d’une ischémie, le contrôle métabolique domine les autres régulations.
Pour suivre la demande en O2 lors d'un effort, la perfusion myocardique périphérique est susceptible d'augmenter de 3 à 5 fois. Cette élévation définit la réserve fonctionnelle: c'est le rapport entre le flux en hyperémie et le flux au repos. Ce rapport est sérieusement abaissé en présence d'une sténose coronarienne, car le flux maximal diminue de manière beaucoup plus importante que le flux au repos. La signification fonctionnelle d'une sténose est ainsi différente du degré de rétrécissement dans près de la moitié des lésions coronaires stables [19]. A vasodilatation maximale, le flux devient pression dépendant et la courbe devient une droite (Figure 5.133). La pente de cette droite est diminuée par la coronaropathie, par la tachycardie et par une augmentation de la contractilité ou de la pression télédiastolique du ventricule gauche; dans ces conditions, la réserve coronarienne est abaissée. Lors d'anémie, d'hypertrophie ventriculaire ou d'effet inotrope positif, la courbe d'autorégulation est déplacée vers le haut; dans ce cas, la réserve coronarienne est également diminuée. Une augmentation du flux coronaire au-delà du seuil d'autorégulation entraîne un effet inotrope positif [29].
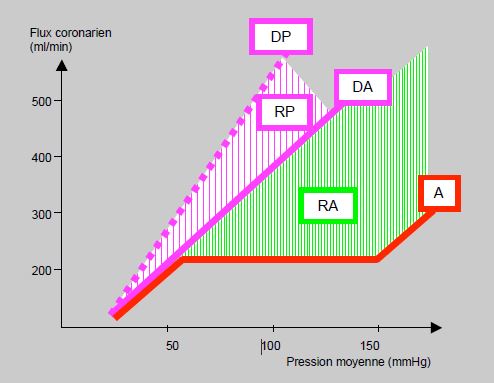
Figure 5.133 : Variation du flux coronaire en fonction de la pression de perfusion lors de vasodilatation maximale. A: courbe d'autorégulation; le flux est stable entre 60 et 160 mmHg de pression aortique. DA: courbe à vasodilatation physiologique maximale; le flux est pression-dépendant. RA: réserve coronarienne de l'autorégulation. DP: courbe à vasodilatation pharmacologique maximale. RP: réserve coronarienne supplémentaire due à la vasodilatation pharmacologique [13].
La fraction de flux de réserve (FFR, fractional flow reserve) est le rapport entre le flux dans une artère sténotique et le flux dans la même artère en vasodilatation maximale. Sa valeur normale est > 1 ; plus la sténose est serrée, plus le rapport diminue (Voir Chapitre 09, Investigations coronarienne). C’est un index spécifique de la sévérité fonctionnelle des sténoses épicardiques. En coronarographie, il est beaucoup plus facile de mesurer la pression que le flux, raison pour laquelle on utilise le rapport entre la pression aortique et la pression moyenne distale à la lésion (mesurée par le cathéter glissé au travers de la sténose) : Pdist / Pao [20]. Pour minimiser l’effet de la résistance artériolaire périphérique, on produit une hyperémie maximale par administration de vasodilatateur intracoronarien (adénosine, papavérine, nicorandil) et l’on enregistre la mesure au nadir de la Pdist [35]. Un index < 0.8 est actuellement considéré comme un critère d’ischémie active et une indication à la revascularisation. Comparée aux indications anatomiques de l'angiographie chez les polytronculaires (sténose > 70%), l’évaluation de la FFR permet de mieux cibler les vaisseaux à revasculariser [3].
La régulation autonome
Le contrôle neuro-humoral (ganglion stellaire et chaîne sympathique D1-D4, nerf vague) concerne les vaisseaux de grand et moyen diamètres [9].
- Les récepteurs α1 prédominent dans les vaisseaux épicardiques de capacitance, dont la vasoconstriction tend à contrecarrer le reflux du à la compression systolique des vaisseaux intramuraux et sous-endocardiques.
- Les récepteurs α2 sont présents surtout dans les vaisseaux de ≤ 1.0 mm et commandent une vasodilatation par l’intermédiaire du NO•.
- Les récepteurs β1 et β2 sont prépondérants dans les vaisseaux résistifs, où leur stimulation provoque une vasodilatation.
- Certaines facteurs hormonaux sont vasoconstricteurs (endothéline, angiotensine II, vasopressine, thromboxane), d’autres vasodilatateurs (prostaglandines I2).
Ainsi la vasoconstriction sympathique tamponne l'aspect biphasique du flux; elle préserve le flux sous-endocardique et la vasodilatation distale β prévient une baisse importante de la PO2 tissulaire [25]. En cas de β-blocage, une stimulation sympathique se traduira seulement par son effet α, d'où un risque de spasme coronarien; c'est la raison pour laquelle les β-bloqueurs sont contre-indiqués dans l'angor de Prinzmetal. Par contre, ces mêmes β-bloqueurs ont la capacité d'augmenter la réserve coronarienne: même si le flux au repos est abaissé, le flux à stimulation maximale est augmenté, ce qui est très profitable en cas d'ischémie [5]. Le contrôle neuro-humoral est contre-balancé par l'autorégulation métabolique : une stimulation sympathique conduit à une augmentation de la mVO2, donc à une vasodilatation locale intramyocardique; cet effet tamponne la vasoconstriction épicardique. La bradycardie d'une stimulation vagale baisse la mVO2, donc conduit à une vasoconstriction locale malgré la vasodilatation des artères épicardiques.
La réponse des vaisseaux coronariens artérioscléreux à une stimulation sympathique diffère souvent de celle des vaisseaux sains, parce que l'endothélium est lésé par l'athéromatose et par les dépôts lipidiques. La dysfonction endothéliale empêche l'autorégulation de contrebalancer l'effet α; la réponse peut ainsi consister en une vasoconstriction (alors que des vaisseaux normaux se dilateraient), par exemple lors d'exercice [27,40]. Enfin, les résistances vasculaires coronaires peuvent être considérablement modifiées par la viscosité sanguine. Celle-ci est liée essentiellement au taux de protéines et à l'hématocrite. L'hémoglobine permet le transport d'oxygène, mais la viscosité augmente avec le nombre de globules rouges. Un taux d'Hb de 100 g/L (Ht = 32%) semble le compromis idéal entre le transport d'O2 maximal et la viscosité la plus faible [28]. Le myocarde des deux ventricules tolère un Ht de 20% sans souffrir dans son apport d'O2 [11].
L’endothélium vasculaire
L'endothélium des coronaires, comme celui des autres vaisseaux, n'est pas qu'un simple revêtement antithrombogène. Il réagit à des agents pharmacologiques (catécholamines, acétylcholine) et à des stimuli mécaniques (pulsatilité du flux, forces de cisaillement); il sécrète une série de substances vasoactives qui modifient la taille des vaisseaux et leur flux [2,17,36].
- Le monoxyde d'azote (NO•) est le principal agent vasodilatateur distal ; sa sécrétion est stimulée par forces de cisaillement (flux, pulsatilité, pression), mais aussi par l'acétylcholine et la sérotonine [7]; il stimule le GMPc, et provoque une vasodilatation et une baisse de l'adhésivité plaquettaire; il freine la vasoconstriction lorsque le débit baisse [34].
- La prostacycline (PGI2) induit une vasorelaxation par l'intermédiaire de la protéine-kinas A; elle inhibe également l'adhésion plaquettaire [2].
- L'endothéline ET-1 est un vasconstricteur puissant, dont la sécrétion est stimulée par la thrombine, l'angiotensine II, l'adrénaline et la vasopressine [21,38]. Lors de pontages aorto-coronariens, elle est augmentée après la CEC suite à l'ischémie et à la reperfusion [31]. Elle n’est probablement pas impliquée dans la régulation du flux en situation normale.
- La thromboxane A2 est le produit de l'action de la co-oxygénase (COX) sur l'acide arachidonique. Elle provoque une vasoconstriction et une adhésion plaquettaire [17].
- L'hyperpolarisation de la musculature lisse (EDH, endothelium-dependent hyperpolarization) est une activité directe de l'endothélium [36].
Une dysfonction endothéliale apparaît tôt dans l'évolution de la maladie athéromateuse; elle modifie la balance physiologique entre les effets du NO• et ceux de l’endothéline au profit d'une vasocontriction, que la stimulation soit sympathique, parasympathique ou due à l’acidose locale [37]. Ainsi, des stimuli qui entraînent normalement une vasodilatation (hypoxie, exercice et augmentation de la demande en O2, par exemple) peuvent conduire à une vascoconstriciton coronarienne dans des vaisseaux artériosclérotiques [33]. La dysfonction endothéliale pourrait être une origine de l'angor à coronaires saines [1,26]. Associée au stress sympathique et à l'hypercoagulabilité, elle est un des trois principaux éléments dans la genèse des évènements ischémiques périopératoires.
Outre ces agents physiologiques, un certain nombre de substances pharmacologiques agissent sur la régulation du flux coronarien [8].
- Les dérivés nitrés libèrent du NO• ; ils sont vasodilatateurs des vaisseaux de conductance et des collatérales; ils décompriment les vaisseaux sous-endocardiques par baisse de la tension de paroi en diastole (baisse de précharge).
- Les antagonistes calciques dilatent les vaisseaux épicardiques et intramyocardiques.
- Le dipyridamole dilate les vaisseaux intramyocardiques.
- L'adénosine, la papavérine et l'acétylcholine augmentent le flux coronarien par vasodilatation.
- L'ergonovine (ergot de seigle) est un vasoconstricteur utilisé comme test pour l'angor de Prinzmetal.
Les halogénés réduisent la disponibilité en Ca2+ intracellulaire dans la musculature lisse et induisent une vasodilatation systémique. Au niveau de l'endothélium coronarien, ils provoquent une vasoconstriction de très courte durée, mais celle-ci est contre-balancée par une vasorelaxation à long terme indépendante de l'endothélium. Le bilan est une vasodilatation coronarienne [2]. En outre, ils ont un effet protecteur sur les lésions d'ischémie-reperfusion (voir Préconditionnement).
Hormis l'adénosine administrée directement dans les coronaires, toutes ces substances voient leur effet vasodilatateur modifié par l'autorégulation locale (variations de la mVO2) ou par leur activité sur la pression de perfusion systémique. Pour s'assurer qu'une substance soit un coronarodilatateur efficace, il faut maintenir constantes la pression de perfusion systémique et la consommation d'oxygène du myocarde [37].
Particularité du VD
Le travail systolique du VD étant 4 fois plus faible que celui du VG, la perfusion coronarienne du VD présente plusieurs singularités qui la différentient de celle du VG [11].
- La mVO2 droite est la moitié de la gauche (4.5 vs 10 mL/min/100 g).
- L'extraction d'O2 est plus faible (40% au lieu de 70-80%).
- La faible épaisseur et la modeste compression de la paroi droite ne prétéritent pas le flux sous-endocardique.
- Le VD bénéficie d'une collatéralisation depuis le réseau coronarien gauche.
- Le flux coronarien est présent en diastole et en systole (voir Figure 5.116).
- La prédominance de récepteurs α conduit à une vasoconstriction coronarienne lors de stiumlation sympathique.
- La production de NO par l'endothélium est mineure, d'où une faible autorégulation du flux distal en cas de sténose de la CD ou de baisse du flux coronarien.
Si la pression intraventriculaire systolique droite s'élève à cause d'une hypertension pulmonaire, la perfusion systolique est progressivement amputée, et le VD perd près de la moitié de son apport sanguin; il est ischémié malgré la persistance du flux coronarien diastolique (voir Figure 5.117). Il suffit d'une légère hypotension systémique pour que la situation soit critique. Il faut alors augmenter la pression systémique par un vasoconstricteur.
| Flux coronarien |
|
Débit coronaire de base: 220 à 250 mL/min. Le flux est constant pour des PAM de 60 à 160 mmHg; il cesse dans le sous-endocarde si PAM < 50 mmHg. La zone sous-endocardique est la plus à risque d'ischémie. Calcul de la pression de perfusion coronarienne: PPC = PAM – PAPO.
Il existe 3 types de vaisseaux coronariens:
- Epicardiques (< 10% de la résistance totale), régulation endothéliale et neuro-humorale
- Pré-artériolaires (30% de la résistance), contrôle neuro-humoral
- Artériolaire (> 50% de la résistance), autorégulation locale
Le flux coronaire est sous triple régulation:
- Autorégulation métabolique prioritaire (vasodilatation par adénosine, NO•, acidose, hypoxie)
- Régulation neuro-humorale: récepteurs α vasoconstricteurs (vaisseaux épicardiques), récepteurs β vasodilatateurs (artérioles de résistance)
- Facteurs endothéliaux: NO• dilatateur, endothéline constrictrice; endothélium athéromateux: prédominance de réponse vasoconstrictrice
La résistance dans les vaisseaux coronariens est sous le contrôle de: métabolisme local (prioritaire), autorégulation, facteurs neuro-humoraux, facteurs endothéliaux, compression extravasculaire, facteurs rhéologiques (viscosité, Ht idéal 35%).
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2019
Références
- AGEWALL S, BELTRAME JF, REYNOLDS HR, et al. ESC working group position paper on myocardial infarction with non-obstructive coronary arteries. Eur Heart J 2017; 38:143-53
- AGUIRRE JA, LUCCHINETTI E, CLANACHAN AS, et al. Unraveling interactions between anesthetics and the endothelium: update and novel insights. Anesth Analg 2016; 122:330--48
- BAIBHAV B, GEDELA M, MOULTON M, et al. Role of invasive functional assessment in surgical revascularization of coronary artery disease. Circulation 2018; 137:1731-9
- BARON JF, VICAUT E, HOU X, DUVELLEROY M. Independent role of arterial O2 tension in local control of coronary blood flow. Am J Physiol 1990; 258:H1388-94
- BILLINGER M, SEILER C, FLEISCH M, et al. Do beta-adrenergic blocking agents increase coronary flow reserve ? J Am Coll Cardiol 2001; 38:1866-71
- BROTEN TP, FEIGL EO. Role of myocardial oxygen and carbon dioxide in coronary autoregulation. Am J Physiol 1992; 262:H1231-7
- CANTY JM, SCHWARTZ JS. Nitric oxide mediates flow-dependent epicardial coronary vasodilatation to changes in pulse frequency but not mean flow in conscious dogs. Circulation 1994; 89:375-84
- CHENG D, VEGAS A. Anesthesia for the surgical management of ischemic heart disease. In: THYS DM, et al editors. Textbook of Cardiothoracic Anesthesiology. New York, McGraw-Hull Co, 2001, pp 530-88
- CHILIAN WM. Functional distribution of a1 and a2 adrenergic receptors in the coronary microcirculation. Circulation 1991; 84:2108-22
- CHILIAN WM. Coronary microcirculation in health and disease. Summary of an NHLBI workshop. Circulation 1997; 95:522-8
- CRYSTAL GJ, PAGEL PS. Right ventricular perfusion. Physiology and clinical implications. Anesthesiology 2018; 128:202-18
- DAVIES JE, et al. Evidence of a dominant backward-propagating "suction" wave responsible for diastolic coronary filling in humans, attenuated in left ventricular hypertrophy. Circulation 2006; 113:1768-78
- DOLE WP. Autoregulation of the coronary circulation. Progr Cardiovasc Dis 1987; 29:293-323
- DOUCETTE JW, GOTO M, FLYNN AE, et al. Effects of cardiac contraction and cavity pressure on myocardial blood flow. Am J Physiol 1993; 265:H1342-52
- DUNCKER DJ, TRAVERSE JH, ISHIBASHI Y, BACHE RJ. Effect of NO on transmural distribution of blood flow in hypertrophied left ventricle during exercise. Am J Physiol 1999; 276:H1305-12
- FAROUQUE HMO. Effect of ATP-sensitive potassium channel inhibition on resting coronary vascular responses in humans. Circ Res 2002; 90:231-6
- FÉLÉTOU M, HUANG Y, VANHOUTTE PM. Endothelium-mediated control of vascular tone. Br J Pharmacol 2011; 164:894-912
- HOFFMAN JI, SPAAN JA. Pressure flow relations in coronary circulation. Physiol Rev 1990; 70:331-90
- JEREMIAS A, KIRTANE AJ, STONE GW. A test in context. Fractional flow reserve: accuracy, prognostic implications, and limitations. J Am Coll Cardiol 2017; 69:2748-58
- KERN MJ, SAMADY H. Current concepts of integrated coronary physiology in the catheterization laboratory. J Am Coll Cardiol 2010; 55:173-85
- KOUREMBANAS S, MARSDEN PA, MCQUILLAN LP, FALLER DV. Hypoxia induces endothelin gene expression and secretion in cultured human endothelium. J Clin Invest 1991; 88:1054-7
- MARTIN SE, LENHARD SD, SCHMARKEY LS, et al. Adenosine regulates coronary blood flow during increased work ands decreased supply. Am J Physiol 1993; 264:H1438-46
- MERIN RG. Anesthesia and the coronary circulation. Adv Anesth 1989; 6:195-218
- MICHAIL M, DAVIES JE, CAMERON JD, et al. Pathophysiological coronary and microcirculatory flow alterations in aortic stenosis. Nat Rev Cardiol 2018; 15:420-31
- MIYASHIRO JK, FEIGL EO. Feed forward control of coronary blood flow via coronary b-receptor stimulation. Circ Res 1993; 73:252-63
- MOTZ W, VOGT M, RABENAU O, et al. Evidence of endothellial dyfunction in coronary resistance vessels in patients with angina pectoris and nornal coronary angiogram. Am J Cardiol 1991; 68:996-1003
- NABEL EG, SELWYN AP, GANZ P. Paradoxical narrowing of atherosclerotic coronary arteries induced by increases in heart rate. Circulation 1990; 81:850-9
- NATHAN HJM. Coronary physiology. In: KAPLAN JA ed. Cardiac anesthesia. Philadelphia, WB Saunders 1993, 235-60
- OPIE LH. Heart Physiology. From cell to circulation. Philadelphia: Lippincott-Williams & Wilkins, 2004, 648 pp
- O'ROURKE MF, YAGINUMA T, AVOLIO AP. Physiological and pathophysiological implications of ventricular/vascular coupling. Ann Biomed Eng 1984; 12:119-34
- PEARSON PJ, LIN PJ, SCHAFF HV. Production of endothelium-derived contracting factor is enhanced after coronary reperfusion. Ann Thorac Surg 1991; 51:788-93
- ROWELL LB, BRENGELMANN GL, BLACKMON JR, et al. Disparities between aortic and peripheral pulse pressures induced by upright exercise and vasomotor changes in man. Circulation 1968; 37:954-64
- SELWYN AP, KINLAY S, CREAGER M, et al. Cell dysfunction in atherosclerosis and the ischemic manifestations of coronary artery disease. Am J Cardiol 1997; 79:17-23
- SMITH TP, CANTY JM. Modulation of coronary autoregulatory responses by nitric oxide. Evidence for flow dependent resistance adjustments in conscious dogs. Circulation 1993; 73:232-40
- TOTH GG, JOHNSON NP, JEREMIAS A, et al. Standardization of fractional flow reserve measurements. J Am Coll Cardiol 2016; 68:742-53
- TRIGGLE CR, SAMUEL SM, RAVISHANKAR S, et al. The endothelium: influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol 2012; 90:713-38
- VERGROESEN I, KAL JE, VAN WEZEL HB. Coronary vasodilating drug effects or normal coronary blood flow regulation ? J Cardiothorac Vasc Anesth 1998; 12:450-6
- YANAGISAWA M, KURIHARA H, KIMURA S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332:411-5
- ZAUGG M, SCHAUB MC, FOËX P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21-33
- ZHANG C. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res 2003; 92:322-9