La sténose coronarienne est le fait d'une plaque d'athérome à laquelle se surimpose parfois un spasme musculaire. L'athéromatose elle-même est une réponse inflammatoire chronique à un dysfonctionnement endothélial, caractérisé par un taux excessif de protéine C-réactive, et à des stimuli biochimiques comme le cholestérol LDL (Low-density lipoprotins), la fumée ou le diabète. La dysfonction endothéliale est liée à une prédisposition génétique et au stress de paroi (hypertension artérielle). Elle aboutit à l'infiltration, d’abord discrète puis massive, de la paroi vasculaire par des cellules musculaires lisses et par des macrophages remplis de cholestérol à basse densité (foam cells). Il se forme une capsule fibreuse plus ou moins résistante à la rupture, qui isole la lésion du flux sanguin. La dysfonction de l’endothélium s’accompagne d’une perte de sa fonction anticoagulante et vasodilatatrice, et d’une augmentation des déclencheurs de l'adhésivité locale des plaquettes (voir Chapitre 08 Voie cellulaire) [6].
Sténose stable et plaque instable
Classiquement, la maladie coronarienne est divisée en deux catégories, angor stable et syndrome aigu, qui sont liées à l’existence de deux types de plaque athéromateuse (Figure 5.137) [16,17,28,30].
- Plaque athéromateuse stable caractérisée par une partie centrale lipidique de faible dimension, recouverte d'une couche fibro-musculaire épaisse, parfois calcifiée. Elle cause le plus souvent des sténoses serrées (> 75%), bien visibles à l’angiographie, qui croissent de manière progressive et qui limitent le flux sanguin dans tout un territoire.
- Plaque instable, de croissance discontinue et irrégulière, composée d'une partie centrale massive de nature lipidique à haute teneur en cholestérol (LDL), où se mêlent des macrophages, des lymphocytes T et des facteurs tissulaires; cet amas est faiblement encapsulé par une couche fibreuse fine (50-65 μm) qui présente des signes d’érosion et de cicatrisation. A l’angiographie, cette plaque ne se manifeste que par une sténose modeste (≤ 50%) et non-limitative du flux, car elle tend à bomber vers l’extérieur et non vers l’intérieur de l’artère [29]. Son risque est tributaire de son activité inflammatoire et de sa susceptibilité à la rupture, mais non du degré de sténose qu’elle occasionne [8,18,23].
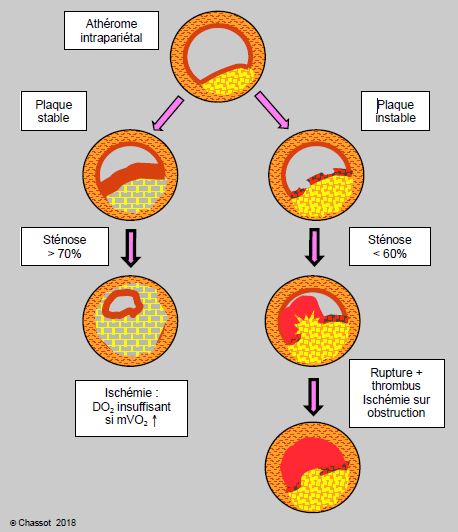
Figure 5.137 : Evolutions possibles d'une plaque athéromateuse à partir d'un noyau lipidique encapsulé par du tissu fibreux (fibro-athérome). La lésion stable est riche en zones calcifiées, en fibre musculaire lisse et en collagène; elle est recouverte d'une capsule fibreuse résistante. La lésion instable a perdu ses fibres conjonctives et musculaires lisses; elle est remplie de matériel lipidique et de macrophages, sa fine capsule fibreuse est très inflammatoire. Sa rupture est potentialisée par les forces de cisaillement et le syndrome inflammatoire. La rupture de la plaque met le matériel lipidique et les éléments cellulaires en contact avec les facteurs de coagulation et les thrombocytes circulants. Un thrombus se forme. L'évolution peut se faire vers l'occlusion totale du vaisseau et l'infarctus, ou vers une fibrinolyse spontanée et une cicatrisation. Le thrombus peut secondairement se recanaliser. L'étendue de l'infarctus dépend du territoire perfusé par le vaisseau occlus, de son degré de collatéralisation, de la demande en O2 du myocarde au moment de l'accident évolutif, et des facteurs induisant la fibrinolyse et la recanalisation; l'état d'hypercoagulabilité périopératoire agrave évidemment la situation.
La fragile capsule fibreuse d’une plaque instable est tissée de collagène synthétisé par les cellules musculaires lisses. Les macrophages, stimulés par les cellules T activées lors d’une réaction inflammatoire, sécrètent des protéinases qui dégradent ce collagène. La rupture subséquente de la capsule met à nu le matériel lipidique et les macrophages, qui entrent alors en contact avec le sang. La violente réaction inflammatoire qui accompagne un infarctus ou une opération chirurgicale déstabilise les plaques sur tout l’arbre coronarien, ce qui explique la fréquence des syndromes coronariens aigus survenant à la suite de ces évènements et le rôle protecteur d’une revascularisation précoce [14]. Lorsque la capsule est fissurée, le facteur tissulaire (FT) présent sur les macrophages et les cellules musculaires lisses entre en contact avec le facteur VII circulant et déclenche la formation de thrombine (voir Figure 8.3). D’autre part, le facteur von Willebrand, lui aussi mis à nu par la rupture, immobilise et active les plaquettes en circulation (voir Figure 8.5) [29]. L'adhésivité de ces dernières est stimulée et conduit à une thrombose locale; celle-ci peut être spontanément lysée, emboliser en périphérie, se recanaliser, ou occlure totalement le vaisseau. Le résultat est un syndrome coronarien aigu ou un infarctus. Les plaquettes stimulées induisent la libération de substances puissamment vasoconstrictrices (thromboxane A2, sérotonine, endothéline).
Cette description a été affinée ces dernières années avec l'étude plus approfondie du rôle des plaques instables dans les syndromes coronariens aigus [2]. En effet, il s'est avéré que la plupart des plaques à fine capsule restent stables et que seuls 5% d'entre elles causent un SCA dans les 3 ans; par ailleurs, 60% des SCA survenus ultérieurement ont été déclenchés par des lésions différentes de celles incriminées au départ (culprit-lesion) [45]. D'autre part, près de la moitié des SCA ne s'accompagne pas de réaction inflammatoire systémique (CRP normale) [12]. De plus, 20% des SCA surviennent en l'absence de thrombose coronarienne [36]. On distingue donc maintenant cinq situations différentes pouvant toutes conduire à un syndrome coronarien aigu (SCA) (Figure 5.138) [12,24].
- Rupture de plaque instable avec réaction inflammatoire systémique; c'est l'image classique déjà décrite ci-dessus. Présente dans 60% des cas de SCA, elle est très liée aux facteurs de risque cardiovasculaire. La fine capsule qui recouvre le dépôt lipidique se rompt sous l'action d'agents inflammatoires; la zone est infiltrée de monocytes et la CRP est élevée.
- Rupture de plaque instable sans réaction inflammatoire; le stress émotionnel, l'hyperactivité sympathique et les états d'hypercoagulabilité sont impliqués dans l'étiologie de cette forme particulière de rupture de plaque instable (5% des cas de SCA).
- Erosion de l'endothélium par apoptose avec perte de cellules endothéliales et infiltration de neutrophiles mais sans implication inflammatoire (30% des cas de SCA); elle est fréquemment associée à l'hypertriglycéridémie, au diabète, à l'âge et au sexe féminin [39]. Les points d'endothélium ainsi dénudés sont des attracteurs pour les plaquettes et les poly-morphonucléaires; il se forme un "thrombus blanc" riche en thrombocytes qui active la thrombine et le dépôt local de fibrine. Cette lésion est le plus souvent liée à un infarctus sans sus-décalage ST (non-STEMI). Il est probable que la thrombectomie simple, les antiplaquettaires, les anticoagulants et les statines soient plus efficaces que la revascularisation sur ce type de lésion [42]. Le diagnostic différentiel de l'érosion endothéliale peut se faire au moyen de la tomographie par cohérence optique (optical coherence tomography, imagerie infra-rouge de la paroi vasculaire par cathétérisme intra-coronarien).
- Vasospasme épicardique ou microcirculatoire; ce mécanisme, lié à un défaut de production locale de NO, est impliqué dans les SCA sans lésion angiographique significative comme le Takotsubo ou l'angor instable (5% des cas de SCA). Il concerne plus fréquemment les femmes que les hommes [39]. Le diamètre du vaisseau est réduit de > 90%. Le spasme peut être déclenché par l'ergonovine ou l'acétylcholine chez 49% des patients qui présentent un SCA sans lésion coronarienne appréciable [37]. La thérapeutique comprend des vasodilatateurs: bloqueurs calciques, dérivés nitrés, fasudil.
- Dissection spontanée d'une artère coronaire; cette affection rare touche plutôt des femmes avant la ménopause qui ne présentent pas de facteur de risque particulier. Le traitement est de préférence conservateur (anticoagulant), car l'angioplastie est associée à un haut degré d'échec technique.
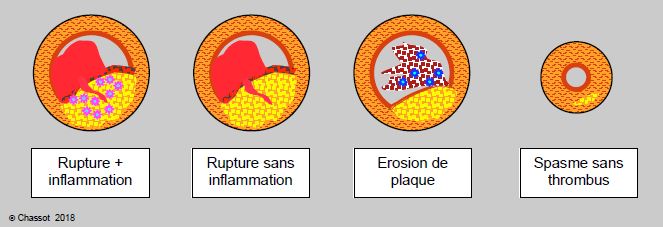
Figure 5.138: Mécanismes étiologiques du syndrome coronarien aigu. On en distingue 4 types: rupture de plaque instable avec réaction inflammatoire majeure et infiltration de monocytes (en violet), rupture de plaque instable sans inflammation, simple érosion de l'endothélium avec thrombus blanc riche en plaquettes et en leucocytes (en bleu), et vasospasme sans thrombus affectant les vaisseaux épicardique et/ou la microcirculation [12].
La pathologie coronarienne se révèle très systémique et très changeante en l'espace de quelques mois. Dans un suivi d'une année par ultrasons intracoronariens, 75% des plaques instables se sont recouvertes d'une capsule épaisse et ont perdu leur caractère d'instabilité, et le 25% restant n'a occasionné aucun SCA pendant la période d'observation [27]. Les lésions cicatricielles sont ré-endothélialisées, épaissies, et contiennent de multiples couches de tissu nécrotique et de tissu fibreux avec plus ou moins de calcifications [38]. La rupture de plaque est fréquente en-dehors de la zone incriminée dans un SCA, mais elle est le plus souvent silencieuse. Pour se déclencher, l'occlusion vasculaire réclame en plus un état thrombotique et hypercoagulatoire qui est influencé par de nombreux phénomènes. Le risque coronarien est donc largement multifactoriel et dépend davantage de l'étendue de la maladie athéromateuse que des lésions individuelles; il est plus directement lié à l'importance de l'artériosclérose, au status inflammatoire et à l'état pré-thrombotique qu'au degré de sténose ou d'instabilité des lésions détectées [2].
Ces types de plaques conduisent schématiquement à trois modalités de pathologies ischémiques [20,29,31].
- La plaque stable est responsable d'épisodes d'angor itératifs, caractéristiques d’un déséquilibre entre l’apport et la demande en O2 : tachycardie, hypertension, douleur, stress, exercice (demand ischaemia). Elle se caractérise par des sténoses serrées (> 75%) à l’angiographie et par des épreuves d’effort en général positives. Lorsqu'elle croît jusqu'à l'occlusion, cette sténose donne lieu à un infarctus de type non-Q, accompagné de sous-décalage du segment ST, correspondant à une lésion sous-endocardique. Il survient fréquemment chez des malades âgés, diabétiques, insuffisants cardiaques, ou souffrant d'angor avec modifications du segment ST et ancien infarctus à l'ECG [20]. Son étendue est inversement proportionnelle au degré de collatéralisation. La meilleure prévention contre cet accident est le β-blocage. Un bas débit (hypotension sévère), une viscosité élevée (déshydratation) et/ou une vasoconstriction locale (endothéline) peuvent parfois causer une thrombose aiguë sur une plaque stable lorsque la sténose est très serrée [18].
- La rupture de plaque instable est sans relation particulière avec une demande accrue en O2, mais elle est responsable de la grande majorité des syndromes coronariens aigus [50]. Elle ne produit que des sténoses modestes à l’angiographie (< 60%), et laisse les tests d’effort le plus souvent silencieux [41]. Sa thrombose cause une occlusion du vaisseau (supply ischaemia) et un infarctus transmural, accompagné de surélévation du segment ST et d'onde Q dans 75% des cas [1]. Elle est responsable de la majorité des syndromes coronariens aigus et des morts subites. L’infarctus survient dans des régions dépendant de vaisseaux peu sténosés à la coronarographie [19] : 80% des lésions coronariennes en relation avec ce type d'infarctus ne sont pas considérées comme significatives lors d'un examen préopératoire [11,13,15]. La prévention est surtout liée aux antiplaquettaires et aux statines [9,10]. Ces dernières ont un effet stabilisateur sur l’équilibre des plaques athéromateuses qui s’étend au-delà de leur capacité à abaisser les lipides circulants [21].
- La résorption du thrombus accumulé sur la plaque instable et la recanalisation spontanée de la lumière vasculaire se manifestent par un épisode d’angor instable sans surélévation du segment ST [5,30]. Des épisodes répétitifs peuvent déformer progressivement la paroi vasculaire jusqu’au moment où un thrombus devient occlusif et cause un infarctus.
Alors qu’elle est la cause de plus des deux tiers des syndromes coronariens aigus en clinique, la rupture de plaque instable n’est à l’origine que de la petite moitié des infarctus postopératoires, où l’ischémie par déséquilibre DO2/VO2 est plus fréquente [28]. Les infarctus périopératoires présentent moins souvent un thrombus intracoronarien que les infarctus rencontrés en cardiologie (13% versus 67% des cas) [44]. Ceci traduit l'implication plus fréquente d'une altération du rapport DO2/VO2 dans la genèse des premiers que dans celle des seconds, comme le prouve le fait qu'une hypotension peropératoire (baisse de la PA de ≥ 40% par rapport à la valeur pré-induction pendant ≥ 30 minutes) double le risque de lésion myocardique [48]. Le sort lié à un thrombus dépend de la durée et du degré d’occlusion de la coronaire. Dans un vaisseau terminal, une interruption du flux de plus de 20 minutes entraîne la nécrose des tissus. La balance entre thrombolyse et facteurs procoagulants, la réaction inflammatoire systémique, les conditions de flux, la géométrie de la lésion, l’éventuelle collatéralisation, et le degré de vasodilatation / vasoconstriction locale décident du destin du myocarde distal à la lésion.
Le patient vulnérable
La vulnérabilité d’un patient à un accident cardiovasculaire aigu (ischémie, infarctus ou mort subite) est basée sur quatre phénomènes : l’importance de la sténose coronarienne (sténose critique), le degré d’instabilité des plaques athéromateuses, la susceptibilité du myocarde et les facteurs déséquilibrants humoraux [34,35].
- Une sténose stable critique est caractérisée par son haut degré d’obstruction au flux (> 90%), sa localisation sur un gros tronc (tronc commun, IVA ou CX proximales), sa faible collatéralisation et l’importance du territoire qu’elle vascularise.
- Une plaque instable est un phénomène dynamique dont l'évolution n'est pas prédictible [8]. De nombreux facteurs interviennent pour en accroître le risque.
- Facteurs structurels : grande taille de la masse centrale lipidique, fragilité de la capsule susceptible de se rupturer, nombre élevé de macrophages et pauvreté en cellules musculaires lisses.
- Facteurs mécaniques : le mince recouvrement endothélial est facilement endommagé et rompu par les forces de cisaillement liées à une poussée hypertensive, à la tachycardie, au spasme coronarien ou à un flux turbulent.
- Facteurs humoraux : le syndrome inflammatoire, le diabète, la nicotine et l'hypercholestérolémie déséquilibrent la masse lipidique.
- Facteurs génétiques : certains malades développent des plaques fragiles alors que d'autres résistent à l'apport de cholestérol ; certaines populations sont particulièrement sensibles aux effets de la diète.
- Le myocarde module les effets de l’ischémie selon une série d’éléments.
- Susceptibilité aux arythmies malignes.
- Etendue et l’emplacement du territoire concerné.
- Degré de collatéralisation.
- Degré de la stimulation sympathique.
- Effet de préconditionnement par les épisodes déjà subis.
- La susceptibilité à la rupture et à la thrombose est liée à plusieurs éléments systémiques véhiculés par le sang [4,23].
- Anomalies coagulatoires ; les syndromes coronariens aigus sont associés à une élévation du fibrinogène et des inhibiteurs du plasminogène ; la période opératoire est caractérisée par une augmentation de l’adhésivité plaquettaire et une baisse de la fibrinolyse.
- Facteurs inflammatoires : de nombreux facteurs sériques prédisent le risque de complications cardiovasculaires : interleukine-6, protéine C-réactive (CRP) ; le syndrome inflammatoire systémique accompagnant la chirurgie est un élément de fort déséquilibre pour les plaques instables [43].
- Facteurs humoraux : taux élevé de catécholamines et de thromboxane A2 (vasoconstricteur libéré par les plaquettes activées), hyperviscosité.
- Activation globale : on a démontré que les syndromes coronariens aigus sont fréquemment associés à une activation générale de toutes les plaques coronariennes, et à la rupture de nombreuses d’entre elles ; le plus souvent, une seule est l’objet d’une thrombose massive qui provoque un infarctus [7,32].
Dans la période postopératoire, la collusion du stress, des variations hémodynamiques, de la forte demande en O2, de la thrombogénécité augmentée et du syndrome inflammatoire chirurgical sont un cocktail particulièrement explosif pour des plaques instables. C’est la raison pour laquelle la protection myocardique (β-bloqueur, antiplaquettaires, statines) doit être maximale chez les patients à risque.
| Sténose coronarienne (I) |
|
Il existe schématiquement 2 types de plaque athéromateuse dans l'arbre coronarien.
La plaque stable: partie lipidique enfouie dans du tissu cicatriciel, endothélium épaissi, calcifications.
- Sténose serrée (> 75%) ne permettant pas un DO2 suffisant si ↑ mVO2, angor d'effort (demand ischaemia);
- Visible à l'angiographie, test d'effort le plus souvent positif
- Conduit à un infarctus non-Q avec sous-décalage ST (non-STEMI)
- Responsable de 60% des infarctus postopératoires (pic d'incidence: 3ème jour postop)
- Prophylaxie: ↑ DO2, ↓ mVO2, β-bloqueurs
La plaque instable: accumulation lipidique avec macrophages et réaction inflammatoire, recouverte d'une capsule mince et friable.
- Sténose peu serrée (< 60%), ne compromettant pas le DO2; angor rare
- Sténose non-significative à l'angiographie, test d'effort le plus souvent négatif
- Instabilité, rupture et thrombose du vaisseau (supply ischaemia)
- Conduit à un infarctus avec onde Q et sus-décalage ST (STEMI)
- Responsable de 40% des infarctus postopératoires (< 36 heures postop)
- Prophylaxie: antiplaquettaires, statines
Cinq situations peuvent conduire à un syndrome coronarien aigu:
- Rupture de plaque instable avec réaction inflammatoire systémique
- Rupture de plaque instable sans réaction inflammatoire systémique
- Erosion de l'endothélium
- Vasospasme coronarien
- Dissection spontanée d'une coronaire
La vulnérabilité à l'ischémie est déterminée par:
- Degré d'obstruction au flux d'une sténose stable serrée
- Degré d'instabilité d'une plaque inflammatoire instable
- Etendue et localisation du myocarde ischémié
- Facteurs humoraux (agrégabilité plaquettaire, syndrome inflammatoire, stress)
|
Effet hémodynamique d’une sténose
Une sténose devient hémodynamiquement significative au-delà de 70% de rétrécissement. En aval d'une sténose, le lit vasculaire est dilaté pour maintenir le flux maximal. Une sténose est dite critique lorsqu'elle induit la dilatation maximale possible dans le réseau distal; toute réduction supplémentaire du diamètre entraîne une baisse du flux. Lorsque la réserve coronarienne est atteinte (sténose de > 85%), l'autorégulation est perdue et le flux devient dépendant de la pression (voir Figure 5.132 et Figure 5.133). Lorsque l'endothélium est lésé par l'athéromatose, l'hypercholestérolémie, le diabète ou l'hypertension, la réserve coronarienne est perdue parce que l'autorégulation ne fonctionne plus normalement et la réponse à une stimulation sympathique devient exagérée [33].
Au niveau d'une sténose, un fluide transforme une partie de son énergie potentielle en énergie cinétique. Il y a accélération mais perte de charge: la pression post-sténotique est plus basse que la pression pré-sténotique. La chute de pression est directement proportionnelle à la longueur de la sténose, mais varie inversément à la puissance 4 par rapport au diamètre de la lumière (loi de Poiseuille). La relation entre le flux à travers une sténose et le gradient de pression engendré n'est pas linéaire, mais croît exponentiellement (Figure 5.139) [26]. La résistance et la baisse de flux ne deviennent significatives qu'à partir de 70% de rétrécissement; en dessous de 50%, les variations sont négligeables. Lorsqu'une sténose augmente de 80 à 90%, la résistance augmente trois fois. Les sténoses modérées n'ont donc pas d'effet significatif sur le flux. Le gradient de pression varie avec le carré de la vitesse du flux (loi de Bernoulli); l'effet d'une sténose augmente donc exponentiellement avec le débit sanguin; il est plus marqué à l'effort qu'au repos. Mais le flux ne dépend pas que du diamètre de la sténose. Il est encore tributaire de sa longueur, de sa géométrie, de son degré d'excentricité, et de facteurs rhéologiques du sang comme l'hématocrite ou le taux de protéines plasmatiques [22].
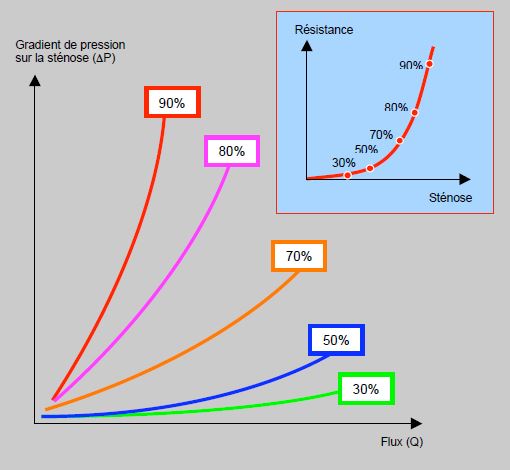
Figure 5.139 : Relation entre le flux et le gradient de pression à travers une sténose. Les courbes illustrent la relation pour différents degrés de rétrécissement dans un vaisseau de 3 mm de diamètre. En cartouche, la courbe démontre la variation de la résistance créée par la sténose en fonction du degré de rétrécissement de celle-ci. Les courbes sont exponentielles; les variations sont négligeables en-dessous de 50%, faibles jusqu'à 70%, et ne deviennent significatives qu'à 75% et au-delà. En effet, la résistance au fluide dans un tube est fonction de la quatrième puissance de son rayon (loi de Poiseuille) [26].
La fraction de réserve de flux est le rapport entre le flux dans une artère sténotique et le flux dans la même artère en vasodilatation maximale. Sa valeur normale est > 1 ; plus la sténose est serrée, plus le rapport diminue. Un rapport de 0.75 signifie que le vaisseau sténosé ne fournit que le 75% du flux en périphérie. C’est un index spécifique des sténoses épicardiques. En coronarographie, il est beaucoup plus facile de mesurer la pression que le flux, raison pour laquelle on utilise le rapport entre la pression aortique et la pression moyenne distale à la lésion (mesurée par le cathéter intracoronarien) : Pdist/Pao [25]. Pour minimiser l’effet de la résistance artériolaire périphérique, on produit une hyperémie maximale par administration de vasodilatateur (adénosine, papavérine) et l’on enregistre la mesure au nadir de la Pdist [47]. Un index < 0.8 est bien corrélé à la positivité des tests d’effort ; cette valeur est considérée comme un critère d’ischémie active et une indication à la revascularisation [3].
Une sténose peut être dynamique: le vasospasme épicardique (angor de Prinzmetal) est l'exemple typique de l'ischémie induite par une vasoconstriction active; il se traduit par une ischémie transmurale avec élévation du segment ST [40]. D'autre part, une sténose athéromateuse est rarement circulaire, et comporte une partie musculaire résiduelle qui peut se spasmer suffisamment pour rendre la sténose critique et occasionner une ischémie en aval. Ce spasme est dû à la libération locale de substances vasoconstrictrices par les plaquettes adhérant à la sténose (sérotonine, thrombexane A2), et par le déséquilibre de l'endothélium lésé qui présente une prédominance d'effets vasoconstricteurs (endothéline, angiotensine II) [49]. La fumée du tabac favorise ce déséquilibre, donc le spasme coronarien [46].
Facteurs aggravants
L’effet clinique d’une sténose coronarienne dépend de plusieurs phénomènes.
- Localisation : ischémie du nœud atrio-ventriculaire (bloc complet), système gauche-dominant ou droite-dominant.
- Etendue de myocarde ischémié : les sténoses ostiales, les sténoses du tronc commun ou leurs équivalents (IVA proximale, CX proximale) réduisent le flux d’une grande masse myocardique dans des territoires dont la contribution à l’éjection systolique est majeure.
- Degré de collatéralisation permettant de court-circuiter la lésion sténosante.
- Lésions micro-angiopathiques diabétiques (épaississement de la membrane basale des capillaires qui limite la diffusion de l’O2).
- Augmentation de la demande en O2.
| Sténose coronarienne (II) |
|
Une sténose est considérée comme significative à > 70% de rétrécissement de la lumière. Distalement à la sténose, le réseau est vasodilaté et le flux pression-dépendant.
Le flux distal baisse exponentiellement avec le degré de sténose. Le gradient de pression augmente avec le carré de la vélocité du flux; il est proportionnel à la puissance 4 du diamètre de la sténose.
La fraction de réserve de flux est le rapport entre la pression aortique et la pression moyenne distale à la lésion; il est normalement > 1. Un rapport < 0.8 est une indication à la revascularisation.
L'effet d'une sténose dépend aussi de sa localisation, de sa longueur, de sa géométrie, du degré de collatéralisation, du flux distal et de la micro-angiopathie.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- ANTMAN EM, BRAUNWALD E. ST-elevation myocardial infarction: Pathology, pathophysiology, and clinical features. In: ZIPES DP, et al, eds. Braunwald's heart disease. A textbook of cardiovascular medicine, 7th edition. Philadelphia, Elsevier-Saunders, 2005, 1141-65
- ARBAB-ZADEH A, FUSTER V. The myth of "vulnerable plaque". Transitioning from a focus on individual lesions tp atherosclerotic disease burden for coronary artery disease risk assessment. J Am Coll Cardiol 2015; 65:846-55
- BAIBHAV B, GEDELA M, MOULTON M, et al. Role of invasive functional assessment in surgical revascularization of coronary artery disease. Circulation 2018; 137:1731-9
- BLAKE GJ, RIDKER PM. Inflammatory biomarkers and cardiovascular risk prediction. J Intern Med 2002; 252:283-94
- BORISSOFF JI, SPRONK HMH, TEN CATE H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med 2011; 364:1746-60
- BRUCKNER R, MUGGE A, SCHOLZ H. Existence and functional role of alpha1-adrenoreceptors in the mammalian heart. J Mol Cell Cardiol 1985; 17:639-45
- BUCHALTER MB, WEISS JL, ROGERS WG, et al. Noninvasive quantification of left ventricular rotational deformation in normal humans using magnetic resonance imaging myocardial tagging. Circulation 1990; 81:1236-44
- CASSCELLS W, NAGHAVI M, WILLERSON JT. Vulnerable atherosclerotic plaque. A multifocal disease. Circulation 2003; 107:2072-5
- CHASSOT PG, DELABAYS A, SPAHN DR. Perioperative use of antiplatelet drugs. Best Pract Res Clin Anesthesiol 2007; 21:241-56
- CHASSOT PG, DELABAYS A, SPAHN DR. Perioperative antiplatelet therapy: The case for continuing therapy in patients at risk of myocardial infarction. Brit J Anaesth 2007; 99:316-28
- COHEN MC, ARETZ TH. Histological analysis of coronary artery lesions in fatal postoperative myocardial infarction. Cardiovasc Pathol 1999; 8:133-9
- CREA F, LIBBY P. Acute coronary syndromes. The way forward from mechanisms to precision treatment. Circulation 2017; 136:1155-66
- DAWOOD MM, GUPTA DK, SOUTHERN J, et al. Pathology of fatal perioperative myocardial infarction: implications regarding physiopathology and prevention. Int J Cardiol 1996; 57:35-44
- DUTTA P, COURTIES G, WEI Y, et al. Myocardial infarction accelerates atherosclerosis. Nature 2012; 487:325-9
- ELLIS SG, HERTZER NR, YOUNG JR, et al. Angiographic correlates of cardiac death and myocardial infarction complicating major non-thoracic vascular surgery. Am J Cardiol 1996; 77:1126-28
- FUSTER V, BADIMON L, BADIMON J, CHESEBRO JH. The pathogenesis of coronary artery disease and the acute coronary syndromes. N Engl J Med 1992; 326:242-50
- FUSTER V, BADIMON L, BADIMON J, CHESEBRO JH. The pathogenesis of coronary artery disease and the acute coronary syndromes. N Engl J Med 1992; 326:310-18
- FUSTER V, FAYAD ZA, BADIMON JJ. Acute coronary syndromes: biology. Lancet 1999; 353(suppl III):5-9
- GIROUD D, LI JM, URBAN P, et al. Relation of the site of acute myocardial infarction to the most severe coronary arterial stenosis at prior angiography. Am J Cardiol 1992; 69:729-32
- GUADANGNOLI E, HAUPTMAN PJ, AYANIAN JK. et al. Variation in the use of cardiac procedures after acute myocardial infarction. N Engl J Med 1995; 333:573-8
- HATTORI K, OZAKI Y, ISMAIL TF, et al. Impact of statin therapy on plaque characteristics as assessed by serial OCT, grayscale and integrated backscatter-IVUS. JACC Cardiovasc Imaging 2012; 5:169-77
- HOFFMAN JI, SPAAN JA. Pressure flow relations in coronary circulation. Physiol Rev 1990; 70:331-90
- HOFFMEISTER HM, HELLER W, SEIPEL L. Activation markers of coagulation and fibrinolysis: alterations and predictive value in acute coronary syndrome. Thromb Haemost 1999; 82:76-9
- KANWAR SS, STONE GW, SINGH M, et al. Acute coronary syndromes without coronary plaque rupture. Nat Rev Cardiol 2016; 13:257-65
- KERN MJ, SAMADY H. Current concepts of integrated coronary physiology in the catheterization laboratory. J Am Coll Cardiol 2010; 55:173-85
- KLOCKE FJ. Measurements of coronary blood flow and degree of stenosis: Current clinical implications and continuing uncertainties. Newsl Council Clin Cardiol AHA 1982(3)
- KUBO T, MAEHARA A, MINTZ GS, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol 2010; 55:1590-7
- LANDESBERG G. The pathophysiology of perioperative myocardial infarction: Facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90-100
- LIBBY P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med 2013; 368:2004-13
- LIBBY P, THEROUX P. Pathophysiology of coronary artery disease. Circulation 2005; 111:3481-8
- MASERI A, FUSTER V. Is there a vulnerable plaque ? Circulation 2003; 107:2068-71
- MAURIELLO A, SANGIORGI C, FRATONI S, et al. Diffuse and active inflammation occurs in both vulnerable and stable plaques of the entire coronary tree: a hisopathological study of patients dying of acute myocardial infarction. J Am Coll Cardiol 2005; 45:1585-93
- NABEL EG, SELWYN AP, GANZ P. Paradoxical narrowing of atherosclerotic coronary arteries induced by increases in heart rate. Circulation 1990; 81:850-9
- NAGHAVI M, LIBBY P, FALK E, et al. From vulnerable plaque to vulnerable patient. A call for new definitions and risk assessment strategies. Part I. Circulation 2003; 108:1664-72
- NAGHAVI M, LIBBY P, FALK E, et al. From vulnerable plaque to vulnerable patient. A call for new definitions and risk assessment strategies. Part II. Circulation 2003; 108:1772-8
- NICCOLI G, MONTONE RA, CATANEO L, et al. Morphological-biohumoral correlations in acute coronary syndromes: pathogenetic implications. Int J Cardiol 2014; 171:463-6
- ONG P, ATHANASIADIS A, HILL S, et al. Coronary artery spasm as a frequent cause of acute coronary syndrome: the CASPAR study. J Am Coll Cardiol 2008; 52:523-7
- OTSUKA F, JONER M, PRATI F, et al. Clinical classification of plaque morphology in coronary disease. Nat Rev Cardiol 2014; 11:379-89
- PAGIDIPATI NJ, PETERSON ED. Acute coronary syndromes in women and men. Nat Rev Cardiol 2016; 13:471-80
- PASUPATHY S, AIR T, DREYER RP, et al. Systematic review of patients presenting with suspected myocardial infarction and nonobstructive coronary arteries. Circulation 2015; 131:861-70
- POLDERMANS D, BOERSMA E BAX JJ, et al. Correlation of location of acute myocardial infarct after non-cardiac vascular surgery with preoperative dobutamine echocardiographic findings. Am J Cardiol 2001; 88:1413-4
- PRATI F, UEMURA S, SOUTEYRAND G, et al. OCT-based diagnosis and management of STEMI associated with intact fibrous cap. JACC Cardiovasc Imaging 2013; 6:283-7
- RIDKER PM, RIFAI N, ROSE L, et al. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediciton of first cardiovascular events. N Engl J Med 2002; 347:1557-65
- SHETH T, NATARAJAN MK, HSIEH V, et al. Incidence of thrombosis in perioperative and non-operative myocardial infarction. Br J Anaesth 2018; 120:725-33
- STONE GW, MAEHARA A, LANSKY AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011; 364:226-35
- SUGIISHI M. Cigarette smoking is a major risk factor for coronary spasm. Circulation 1993; 87:76-9
- TOTH GG, JOHNSON NP, JEREMIAS A, et al. Standardization of fractional flow reserve measurements. J Am Coll Cardiol 2016; 68:742-53
- VAN WAES JA, VAN KLEI WA, WIJEYSUNDERA DN, et al. Association between intraoperative hypotension and myocardial injury after vascular surgery. Anesthesiology 2016; 124:35-44
- ZHANG C. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ Res 2003; 92:322-9
- ZIPES DP, WELLENS HJ. Sudden cardiac death. Circulation 1998; 98:2334-51