Activation de la coagulation
La coagulation est un phénomène local, normalement déclenché par une lésion tissulaire qui rompt la barrière de l’endothélium (pour plus de détails, voir Chapitre 8 Coagulation & hémostase). Quatre processus physiologiques sont en jeu (Figure 7.20) [1,16].
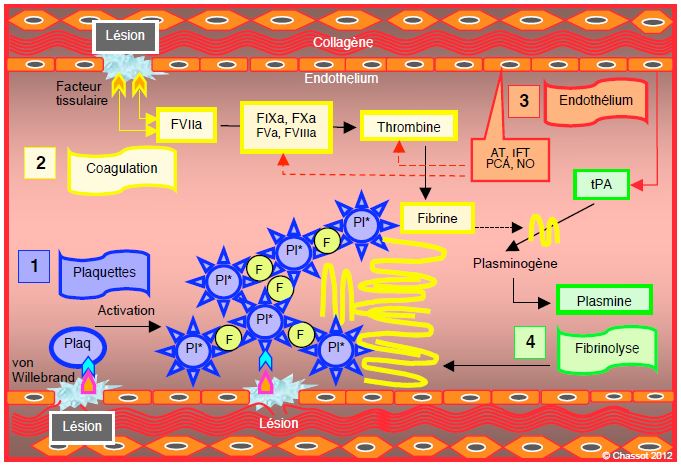
Figure 7.20 : Activation de la cascade coagulatoire lors d’une lésion tissulaire telle que la plaie opératoire. Quatre processus sont en jeu. 1 : bouchon plaquettaire (en bleu). Le facteur von Willebrand est mis au contact des plaquettes par la lésion endothéliale ; celles-ci passent du repos (Plaq) à la phase activée (Pl*). Les plaquettes activées s’ammassent entre elles en se reliant par les attaches de leurs récepteurs GPIIb/IIIa avec le fibrinogène (F). 2 : cascade de la coagulation (en jaune). L’exposition du facteur tissulaire dans la lésion active le Facteur VII circulant en FVIIa, ce qui met en marche la chaîne de la coagulation pour aboutir à la transformation de la prothrombine en thrombine ; celle-ci potentialise la coagulation par rétroaction dans une boucle amplifi-catrice (stimulation des facteurs Va et VIIIa qui à leur tour stimulent les facteurs XIa et Xa). La thrombine transforme le fibrinogène circulant en fibrine ; celle-ci va stabiliser le bouchon plaquettaire (filaments jaunes) et le transformer en thrombus solide grâce à l’action du facteur XIII activé (F XIIIa) qui crée des ponts entre les molécules de fibrine. 3 : les cellules endothéliales normales agissent comme régulateurs (en rouge) pour empêcher la propagation de la coagulation au-delà de la zone lésée. Elles libèrent une série de facteurs inhibiteurs (antithrombine AT, protéine C active PCA, inhibiteur de la voie du facteur tissulaire IFT, NO) qui interrompent la voie de la coagulation. 4 : fibrinolyse (en vert). L’endothélium libère également l’activateur tissulaire du plasminogène (tPA) qui se lie à la fibrine et transforme le plasminogène en plasmine ; cette dernière va lyser la fibrine [25].
- Bouchon plaquettaire. La lésion endothéliale permet un contact entre les facteurs tissulaires (von Willebrand, GPIb/V/IX) et les plaquettes circulantes; celles-ci passent du repos à la phase activée. Les plaquettes activées s’ammassent entre elles en se reliant par les attaches de leurs récepteurs GPIIb/IIIa avec le fibrinogène. Les plaquettes activées et la lésion de l’endothélium fournissent une surface de phospholipides chargés négativement qui est la plateforme sur laquelle se bâtit la cascade de la coagulation.
- Cascade de la coagulation. L’exposition du facteur tissulaire (FT) dans la lésion endothéliale active le Facteur VII circulant en F VIIa, ce qui met en marche la chaîne de la coagulation (voie extrinsèque) pour aboutir à la transformation de la prothrombine (F II) en thrombine (F IIa); celle-ci potentialise l’activation des plaquettes et de la coagulation par rétroaction dans une boucle amplificatrice (stimulation des facteurs Va et VIIIa qui à leur tour stimulent les facteurs XIa et Xa). La thrombine transforme le fibrinogène circulant en fibrine ; celle-ci va stabiliser le bouchon plaquettaire et le transformer en thrombus solide grâce à l’action du facteur XIII activé (F XIIIa) qui crée des ponts entre les molécules de fibrine.
- Les cellules endothéliales normales agissent comme régulateurs pour empècher la propagation des thrombi au-delà de la zone lésée. Elles libèrent une série de facteurs inhibiteurs des plaquettes et de la cascade coagulatoire (antithrombine, NO, inhibiteur de la voie du facteur tissulaire) qui interrompent la voie de la coagulation et la contiennent au site de la lésion.
- Fibrinolyse. L’endothélium libère également l’activateur tissulaire du plasminogène qui se lie à la fibrine et transforme le plasminogène en plasmine ; cette dernière va lyser la fibrine et la rompre en fragments sans activité coagulatoire (D-dimères).
Indépendamment de toute lésion tissulaire, la CEC déclenche directement la formation de thrombine et de fibrine (voie intrinsèque). Cinq minutes après sa mise en route, leur taux est déjà augmenté de 20 fois, alors que ces substances ne se rencontrent normalement qu’au niveau de la plaie et non dans la circulation systémique [5]. Plusieurs phénomènes interviennent [12,25].
- Le système de contact. Au contact de surfaces étrangères chargées négativement comme le verre ou les plastiques, le facteur XII (Hageman) se clive en XIIa (activé) qui transforme la prékallikréine en kallikréine, le facteur XI en XIa et les kininogènes en bradykinine; le taux de cette dernière augmente de 10 fois. L’activation du FXIa aboutit à la formation active de thrombine par la voie intrinsèque de la coagulation. Le facteur XIIa active également la voie du complément et favorise la transformation de plasminogène en plasmine, provoquant la fibrinolyse. Cependant, la balance penche en faveur de l’excès de thrombine et de l’effet procoagulant; cette situation perdure jusqu’au 5ème jour postopératoire [2].
- La voie extrinsèque. L’expression du facteur tissulaire (FT) et la concentration en facteur VIIa sont anormalement élevées en CEC.
- La fibrinolyse. Le taux de plasmine circulante augmente 10-50 fois pendant la CEC; les vitesses de formation et de dégradation de la fibrine étant équivalentes, cette situation revient à une consommation accrue de fibrinogène sans formation de caillots [6].
- Les plaquettes. Elles sont stimulées par le contact avec les surfaces étrangères, par l’héparine et par l’excès de thrombine en circulation; elle adhèrent aux surfaces, y forment des amas et sécrètent de la thromboxane A2 vasoconstrictrice. Leur nombre et leur agrégabilité diminuent de 30-50% au cours d’une CEC, en partie par lésion mécanique dans les pompes et les circuits [24]. Leur fonction est réduite en hypothermie (< 30°C), mais cette dysfonction est réversible au réchauffement; toutefois, elle ne se manifeste pas sur le résultat des tests d’agrégabilité qui sont effectués sur du sang réchauffé à 37°C. La plasmine dissocie le récepteur GP Ib, ce qui active partiellement la plaquette mais la rend moins sensible aux agonistes [9].
- La réaction inflammatoire. Les leucocytes sont activés par les surfaces étrangères; ils vont alors sécréter du facteur tissulaire (FT) qui contribue au développement de la cascade coagulatoire et à la production de thrombine. Les leucocytes activés s'infiltrent entre les cellules endothéliales et produisent des radicaux libres, des superoxydes et des enzymes lysosomiques; c'est la cause de lésions endothéliales, d'augmentation de perméabilité capillaire, d'accumulation liquidienne extracellulaire et de syndrome inflammatoire. Les surfaces étrangères stimulent aussi la voie alternative du complément (la voie classique est déjà activée par le F XIIa); les facteurs C3a et C5a se lient aux leucocytes circulants et contribuent à leur activation.
- L’hémodilution. Les taux de tous les facteurs sont abaissés de 20-30% par la dilution hydrique de la CEC. Les colloïdes abaissent les taux de facteur VIII et von Willebrand; ils freinent l’adhésion plaquettaire [10,24].
- En fin de CEC, le taux de fibrinogène a baissé de 30-40%; le 30% de l’antithrombine III est consommé, ce qui tend à augmenter progressivement la « résistance » à l’héparine. Les facteurs II, VII, IX et X sont diminués de près de 50% après la CEC [11].
- Le sang retransfusé. S’il n’est pas épuré par un système CellSaver™, le sang aspiré dans le péricarde ou le médiastin contient du facteur tissulaire, des complexes thrombine-antithrombine, des activateurs du plasminogène et des déclencheurs inflammatoires. Il contribue massivement aux altérations de la coagulation.
- Hypothermie et acidose. Une coagulopathie s’installe dès 35°C, et la cascade de la coagulation est complètement inactivée à 16°C. L’acidose aggrave la situation et freinant l’activité des facteurs sensibles au pH comme le facteur VIIa.
- Les lésions imparties aux plaquettes et aux facteurs de coagulation (dénaturation protéique) sont directement liées à la durée de CEC, à la profondeur de l'hypothermie (≤ 25°), aux aspirations, et au contact avec l'air (réservoir veineux, aspirations).
L’expression du facteur tissulaire (FT) et de la voie extrinsèque est essentiellement liée au traumatisme chirurgical, à l’activation par les aspirations et à la réaction inflammatoire. Les leucocytes activés s'infiltrent entre les cellules endothéliales et produisent des radicaux libres, des superoxydes et des enzymes lysosomiques; c'est la cause de lésions endothéliales, d'augmentation de perméabilité capillaire, d'accumulation liquidienne extracellulaire et de syndrome inflammatoire systémique (voir Syndrome inflammatoire). Les lésions imparties aux plaquettes et aux facteurs de coagulation (dénaturation protéique) sont directement liées à la durée de CEC, à la profondeur de l'hypothermie (≤ 25°), aux aspirations, et au contact avec l'air (réservoir veineux, aspirations).
On peut réduire l’activation de la coagulation par différents moyens, mais, hormis l’anticoagulation, leur efficacité est très variable.
- Anticoagulation complète par l’héparine non-fractionnée (HNF); l’activité de la thrombine est bloquée lorsque l’ACT est > 480 secondes, à la condition que l’anti-thrombine soit présente en quantité suffisante (voir Héparines). La dose de charge d’héparine pour obtenir une anticoagulation adéquate est de 300-400 UI/kg. L'ACT est contrôlé 3-5 minutes plus tard. Les doses supplémentaires sont titrées selon la réponse individuelle du patient à l’héparine.
- En cas de résistance à l’héparine, supplémentation en anti-thrombine (AT III), car son taux baisse de 40% en CEC à cause de l’hémodilution et de la consommation par l’héparine. Le nadir de la concentration en AT III est atteint au 3ème jour postopératoire [8]. Administration sous forme de concentré d’AT III (500-1’000 UI pour un adulte) ou de plasma frais décongelé (voir ci-après) [31].
- Chez les patients sous anticoagulants ou sous antiplaquettaires, la dose d’héparine utilisée lors de la CEC ou lors d’OPCAB reste la même que la routine habituelle (ACT recherché : > 450 sec et > 300 sec respectivement), car une inhibition incomplète de la thrombine peut conduire à une activation plaquettaire secondaire [4].
- Antifibrinolytiques ; l’acide tranexamique et l’acide amino-caproïque se fixent sur la lysine du plasminogène et bloquent l'activation de la plasmine, donc la fibrinolyse. L’aprotinine est un inhibiteur non spécifique des protéases, qui bloque directement la plasmine (voir Anti-fibrinolytiques).
- Thromboplégie; la CEC et l’héparine activent les plaquettes, qui relâchent leurs granules (ADP, thrombexane), forment des agrégats et adhèrent aux surfaces; 30-50% d’entre elles ne sont plus fonctionnelles en postopératoire et ne réagissent plus à l’ADP ni au collagène [24]. Leur blocage momentané par un agent antagoniste du récepteur P2Y12 (récepteur ADP) comme le cangrelor en perfusion (demi-vie : 9 minutes) les protège de la stimulation et préserve leur fonctionnalité pour le postopératoire [18]. Cette thérapeutique prometteuse est encore en phase d’essai.
- Modifications liées à la technologie de la CEC [12].
- Restriction des aspirations ; le sang récupéré est en contact avec l’air et contient des activateurs de la coagulation (TF, thrombine), de la fibrinolyse (plasmine) et de l’inflammation (interleukines, TNF, C3a, C5a). Les aspirations sont la source principale d'hémolyse, de thrombopénie, de coagulopathie et de stimulation du syndrome inflammatoire [25]. Les perturbations du système coagulatoire sont nettement diminuées lorsqu’on ne recycle pas le sang aspiré ou lorsqu’on le filtre dans un système CellSaver™, mais cette manoeuvre élimine malheureusement les plaquettes, les protéines et les facteurs de coagulation [30].
- Restriction de la taille des circuits; la miniaturisation des circuits et la suppression du réservoir de cardiotomie minimisent le contact du sang avec des surfaces étrangères et suppriment le contact avec l’air, ce qui freine la libération des activateurs de la coagulation et des déclencheurs inflammatoires.
- Biocompatibilité des circuits; les circuits préhéparinés et les circuits imprégnés de polymères particuliers freinent la cascade du complément, l’agrégabilité plaquettaire et l'activation leucocytaire. L’effet clinique est toutefois peu important et se limite à une diminution du taux de FA postopératoire et du temps de séjour aux soins intensifs [20]. La réduction des transfusions n’est pas constante [23].
- Ultrafiltration; la filtration continue en fin de CEC, après la mise en charge (MUF, modified ultrafiltration), permet de réduire l’hémodilution et de soustraire un grand nombre de cytokines et de déclencheurs de la réaction inflammatoire.
- Opération à cœur battant sans CEC ; l’absence de CEC n’élimine pas l’activation coagulo-inflammatoire, mais la réduit; la dysfonction plaquettaire est moindre [28].
L’anticoagulation et la coagulopathie liées à la CEC doivent être respectivement antagonisée et traitée pour limiter les risques hémorragiques. L’antagoniste de l’héparine est la protamine, administrée à raison de 1 mg de protamine pour 1 mg d’héparine (voir Protamine). Un excès de protamine peut inhiber la cascade coagulatoire et l’activité plaquettaire. La protamine est injectée dès la décanulation de CEC. Elle présente plusieurs effets secondaires:
- Vasodilatation systémique et hypotension artérielle;
- Vasoconstriction pulmonaire;
- Réaction antigène-anticorps;
- Réaction anaphylactoïde foudroyante (choc anaphylactique).
| Coagulopathie de la CEC |
|
La coagulopathie de la CEC est liée à 3 phénomènes: l’hémodilution, l’activation et la consommation. La CEC déclenche rapidement la formation de thrombine et de fibrine, indépendamment de toute plaie tissulaire. Cinq systèmes sont activés par le contact avec des surfaces étrangères:
- Le F XII activé déclenche la voie intrinsèque
- Le facteur tissulaire et le facteur VIIa sont augmentés
- Les plaquettes sont stimulées
- L’activation de la fibrinolyse détruit la fibrine formée mais consomme du fibrinogène
- L’activation des leucocytes et du complément déclenche une réponse inflammatoire
systémique massive
Plusieurs moyens sont mis en œuvre pour pallier cette stimulation:
- Anticoagulation complète (héparine 300-400 UI/kg)
- Antifibrinolytique
- Restriction des aspirations, CellSaver™
- Miniaturisation des circuits, circuits biocompatibles
- Ultrafiltration
- Opération à cœur battant
|
Hémodilution
Le mélange du sang avec le liquide d'amorçage (1.0 - 1.5 L de solution hydro-électrolytique) est responsable d'une hémodilution majeure qui abaisse soudainement l'hématocrite aux environs de 25% et qui diminue la pression colloïdo-osmotique de 40% [15]. C'est la cause principale de la chute de pression enregistrée au début de la CEC. La pression remonte ensuite parce que l'hypothermie provoque une stimulation des résistances artérielles périphériques (RAS) et parce que la viscosité augmente à mesure que la température du sang baisse. La chute de la pression osmotique aggrave la fuite liquidienne extracellulaire dans l'espace interstitiel des poumons, du coeur, du foie, des reins, des viscères abdominaux et des muscles.
L'hémodilution est avantageuse sur plusieurs plans (voir Liquide d’amorçage).
- Elle améliore la microcirculation en baissant la viscosité sanguine, ce qui est capital en hypothermie; la viscosité reste stable lorsque l'Ht en pourcent a la même valeur que la température en degrés C°.
- Elle tend à diminuer le besoin en sang allologue et les complications associées à la transfusion.
- Elle est bien tolérée puisque la consommation d'O2 tissulaire est diminuée à froid.
En CEC normothermique, un Ht de 18% suffit juste à remplir les besoins en oxygène d'un malade endormi et curarisé [19]. Lorsque l’Hb est < 70 g/L, le flux sanguin cérébral augmente de 45% et le flux plasmatique rénal s’élève dans la zone corticale, mais la réserve coronarienne diminue de 50% et la perfusion splanchnique est à la limite de l'ischémie [22,26].
L’hémodilution n’est bénéfique que dans certaines limites. Un Ht inférieur à 22%, par exemple, est un facteur prédictif indépendant de morbi-mortalité postopératoire [14]. L'Ht a un impact particulier sur la fonction cérébrale et sur la fonction rénale. Les troubles neurocognitifs deviennent plus importants lorsque l’Ht minimal est de 15-17% [7] .; seul un Ht > 28% assure un status neurologique postopératoire normal [29]. Chez les enfants, le score neurologique et le développement psychomoteur sont meilleurs lorsque l’Ht en CEC est élevé (28%) que lorsqu’il est bas (21%) [17]. D’autre part, la fonction rénale s’aggrave linéairement avec la baisse de l’hémoglobine lorsque l’hématocrite est < 30% [27]. Un Ht de 25-28% en cours de CEC est donc la limite inférieure de sécurité pour garantir la reprise fonctionnelle des organes.
Hémolyse
Le contact avec des surfaces étrangères de différentes natures provoque une série de traumatismes hématologiques plus ou moins sévères, et en général directement proportionnels à la durée de la CEC. De plus, les pompes provoquent des lésions mécaniques des éléments figurés, qui sont fonction de leur degré d'occlusivité et de leur vitesse de rotation. Enfin, les aspirations dans le champ opératoire sont responsables d’une partie des dégâts inflammatoires et érythrocytaires, qui sont d'autant plus graves que les aspirations sont puissantes et prolongées et que l'hémorragie est importante. L'hémolyse qui en résulte est bien visible dans les urines qui deviennent rouge bordeau. Dans ce cas, il est nécessaire de maintenir un débit urinaire satisfaisant et d'alcaliniser les urines avec du bicarbonate de Na+ (50-100 mmoles i.v.) pour freiner la cristallisation de l'Hb libre dans les tubules [13].
Une autre cause d'hémolyse est la présence d'agglutinines froides. C'est une maladie autoimmune caractérisée par la présence d'anticorps causant l'agglutination des érythrocytes en dessous d'un certain seuil de température.
Agglutinines froides
Les agglutinines froides sont des anticorps IgM dirigés contre des antigènes Anti-I présents sur la membranne des globules rouges. Elles causent une agglutination de ces derniers à basse température. Au réchaufffement, ces aggrégats provoquent des thrombi microvasculaires et sont hémolysés, ce qui dégage une grande quantité d'hémoglobine libre. Cette affection est une maladie idiopathique, ou la séquelle d'un processus infectieux ou lymphoprolifératif. Son incidence est inférieure à 1% des patients de chirurgie cardiaque. L'affection se manifeste par des thromboses périphériques et une hémolyse (voir Chapitre 21 Coagulopathies).
Les agglutinines froides sont détectées au test de Coomb direct (présence de complément sur les GR du patient) et indirect (présence d’anticorps sériques). Elles existent chez tous les individus, mais ne réagissent normalement qu’à 0-4°C. Leur signification clinique tient à leur taux sérique et à la valeur de la température à laquelle elles sont activées. Les valeurs considérées comme sûres pour la CEC sont un titre inférieur à 1:32 à une température de 4°C, sans agglutination détectable à 28°C ou au-dessus. Les probabilités de complications peropératoires deviennent significatives pour des taux supérieurs à 1:512 à 4°C, ou inférieurs à cette valeur si la température d'activation est supérieure à 25°C [21].
En salle d'opération, on prend une série de précautions [3].
- Chirurgie en normothermie (CEC > 34°C) ou à cœur battant;
- Réchauffement de la salle d’opération et des perfusions;
- Cardioplégie chaude (> 34°C) cristalloïde ou au sang;
- Réchauffer les poches de sang en cas de transfusion;
- En cas de crise avec hémolyse:
- Réchauffer à 37°C;
- Améliorer la perfusion périphérique avec un vasodilatateur (nitroprussiate);
- Alcaliniser les urines (50-100 mmoles bicarbonate de Na+);
- Méthylprednisolone (500 mg) : efficacité discutée;
- Réduction des taux circulants par plasmaphérèse préopératoire si nécessaire.
Les crises se manifestent par une hémolyse et des occlusions vasculaires périphériques myocardiques, hépatiques et rénales.
| Aspects hématologiques |
|
Le volume d’amorçage de la CEC provoque une hémodilution (Ht 25-28%), nécessaire pour freiner l’augmentation de la viscosité du sang à basse température. En hypothermie, la viscosité reste stable lorsque la valeur de l'Ht en % est la même que celle de la température en degrés C°. Lorsque l’Ht est < 25%, le status neurologique et la fonction rénale postopératoires sont péjorés. Un Ht de 25-28% est la limite inférieure de sécurité pour garantir la reprise fonctionnelle normale des organes.
La CEC provoque une hémolyse, en général infra-clinique. En hypothermie, celle-ci peut devenir massive en présence d’hémagglutinines froides (mises en évidence par un test de Coombs).
La CEC déclenche rapidement la formation de thrombine et de fibrine, indépendamment de toute plaie tissulaire. Quatre systèmes sont activés par le contact avec des surfaces étrangères:
- Le F XII activé déclenche la voie intrinsèque
- Les plaquettes sont stimulées
- La fibrinolyse détruit la fibrine formée mais consomme du fibrinogène
- L’activation des leucocytes et du complément déclenche une réponse inflammatoire
systémique massive
|
© CHASSOT PG, GRONCHI F, Avril 2008, dernière mise à jour, Avril 2018
Références
- ACHNECK HE, SILESHI B, PARIKH A, et al. Pathophysiology of bleeding and clotting in the cardiac surgery patient. Circulation 2010; 122:2068-77
- AVIDAN MS, LEVY JH, SCHOLZ J, et al. A phase III, double-blind, placebo-controlled, multicenter study on the efficacy of recombinant human antithrombin in heparin-resistant patients scheduled to undergo cardiac surgery necessitating cardiopulmonary bypass. Anesthesiology 2005 ; 102 : 276-84
- BRACKEN CA, GURKOWSKI MA, NAPLES JJ, et al. Cardiopulmonary bypass in two patients with previously undetected cold agglutinins. Case Conference. J Cardiothorac Vasc Anesth 1993; 7:743-9
- BROWN C, JOSHI B, FARADAY N, et al. Emergency cardiac surgery in patients with acute coronary syndromes : a review of the evidence and perioperative inmplications of medical and mechanical therapeutics. Anesth Analg 2011 ; 112 : 277-99
- CHANDLER WL, VELAN T. Estimating the rate of thrombin and fibrin generation in vivo during cardiopulmonary bypass. Blood 2003; 101:4355-62
- CHANDLER WL, VELAN T. Plasmin generation and D-dimer formation during cardiopulmonary bypass. Blood Coag Fibrinolysis 2004; 15:583-91
- DE FOE GR, ROSS CS, OLMSTEAD EM, et al. Lowest hematocrit on bypass and adverse outcomes associated with coronary artery bypass grafting. Ann Thorac Surg 2001; 71:769-76
- DIETRICH W, BUSLEY R, SPANNAGL M, et al. The influence of antithrombin substitution on heparin sensitivity and activation of hemostasis during coronary artery bypass graft surgery : a dose-finding study. Anesth Analg 2013 ; 116 :1223-30
- DUNNING J, VERSTEEGH M, FABBRI A, et al. Guidelines on antiplatelet and anticoagulation management in cardiac surgery. Eur J Cardiothorac Surg 2008; 34:73-92
- FRANZ A, BRAUNLICH P, et al. The effects of hydroxyethyl starches of varying molecular weights on platelet function. Anesth Analg 2001 ; 92 : 1402-7
- GHADIMI K, LEVY JH, WELSBY IJ. Prothrombin complex concentrates for bleeding in the perioperative setting. Anesth Analg 2016; 122:1287-300
- GRONCHI F, RANUCCI M. Perioperative coagulation in cardiovascular surgery. In : MARCUCCI C, SCHOETKER P, editors. Perioperative hemostasis. Coagulation for anesthesiologists. Heidelberg : Springer Verlag, 2014, 243-66
- HAASE M, HAASE-FIELITZ A, BELLOMO R, et al. Sodium bicarbonate to prevent increases in serum creatinine after cardiac surgery: a pilot double-blind, randomized controlled trial. Crit Care Med 2009; 37:39-47
- HABIB RH, ZACHARIAS A, SCHWANN TA, et al. Adverse effects of low hematocrit during cardiopulmonary bypass in the adult: should current practice be changed ? J Thorac Cardiovasc Surg 2003; 125:1438-50
- HALL TS. The pathophysiology of cardiopulmonary bypass: The risks and benefits of hemodilution. Chest 1995: 100:88-94
- INNERHOFER P, KIENAST J. Principles of perioperative coagulopathy. Best Pract Res Clin Anaesthesiol 2010; 24:1-14
- JONAS RA, WYPIJ D, ROTH SJ, et al. The influence of hemodilution on outcome after hypothermic cardiopulmonary bypass: results of a randomized trial in infants. J Thorac Cardiovasc Surg 2003; 126:1765-74
- KRAJEWSKI S, KURZ J, NEUMANN B, et al. Short-acting P2Y12 blockade to reduce platelet dysfunction and coagulopathy during experimental extracoroporeal circulation and hypothermia. Br J Anaesth 2012; 108:912-21
- LIAM BL, PLOCHL W, COOK DJ, et al. Hemodilution and whole body balance during normothermic cardiopulmonary bypass. J Cardiothorac vasc Surg 1998; 115:1203-8
- MANGOUSH O, PURKAYASTHA S, HAJ-YAHIA S, et al. Heparin-bonded circuits versus nonheparin-bonded circuits: an evaluation of their effect on clinical outcomes. Eur J Cardiothorac Surg 2007; 31:1058-69
- MONGERO LB, BECK JR (eds). On bypass. Advanced perfusion techniques. Policy and procedure guidelines (CP36): Cold agglutinins. Totowa (NJ, USA): Humana Press 2010, 426-8
- OHRI SK, BOWLES CW, MATHIE RT, et al. Effect of cardiopulmonary bypass perfusion protocols on gut tissue oxygenation and blood flow. Ann Thorac Surg 1997; 64:163-8
- RANUCCI M, BALDUINI A, DITTA A ; et al. A systematic review of biocompatible cardiopulmonary bypass circuits and clinical outcome. Ann Thorac Surg 2009 ; 87 : 1311-9
- ROZENTAL T, SHORE-LESSERSON L. Pharmacologic management of coagulopathy in cardiac surgery: An update. J Cardiothorac Vasc Anesth 2012; 26:660-79
- SNIECINSKI RM, CHANDLER WL. Activation of the hemostatic system during cardiopulmonary bypass. Anesth Analg 2011; 113:1319-33
- SUNGURTEKIN H, COOK DJ, ORSZULAK TA, et al. Cerebral response to hemodilution during hypothermic cardiopulmonary bypass in adults. Anesth Analg 1999; 89:1078-83
- SWAMINATHAN M, PHILIPS-BUTE BG, CONLON PJ, et al. The association of lowest hematocrit during cardiopulmonary bypass with acute renal injury after coronary artery bypass surgery. Ann Thorac Surg 2003; 76:784-91
- UNTCH BR, JESKE WP, SCHWARTZ J, et al. Inflammatory and hemostatic activation in patients undergoing off-pump coronary artery bypass grafting. Clin Appl Thromb Hemost 2008; 14-141-8
- VAN WERMESKERKEN GK, LARDENOYE JWH, HILL SE, et al. Intraoperative physiologic variables and outcome in cardiac surgery. Part II. Neurologic outcome. Ann Thorac Surg 2000; 69:1077-83
- WANG G, BAINBRIDGE D, MARTIN J, et al. The efficacy of an intraoperative cell saver during cardiac surgery: a meta-analysis of randomized trials. Anesth Analg 2009; 109:320-30
- WILLIAMS MR, D’AMBRA AB, BECK JR, et al. A randomized trial of antithrombin concentrate for treatment of heparin resistance. Ann Thorac Surg 2000; 70:873-7