Deux phénomènes particuliers sont liés à l'ischémie, l'hibernation et le préconditionnement (voir Préconditionnement). Quatre autres processus sont liés à la reperfusion myocardique : la sidération, les lésions de reperfusion, la non-reperfusion et les arythmies.
Hibernation
Entre l'ischémie aiguë sans traduction hémodynamique et la nécrose tissulaire de l'infarctus, il existe tout un éventail dans l'intensité et la durée de la dysfonction ventriculaire. Le myocarde hibernant est du tissu viable mais ischémié de manière continue et chronique, qui présente une dysfonction contractile grave et prolongée, sans signes de nécrose, pouvant s'étendre à tout le ventricule [2,19]. Cette situation, liée à une hypoperfusion, est un mode d'auto-préservation rapidement réversible en cas de reperfusion (Figure 5.143) [5]. Le bas débit coronaire suffit à maintenir la viabilité métabolique, mais non la contractilité; le taux d'ATP est normal mais la densité des récepteurs β est diminuée [20]. La fonction peut être stimulée par des agents ionotropes, mais cette stimulation est potentiellement délétère, car l'hibernation est une forme d'autoprotection myocardique [7]. Une revascularisation myocardique peut restaurer la fonction en l’espace de quelques heures à quelques jours [11]. Malgré la mauvaise fonction préopératoire, le pronostic est excellent si la viabilité du myocarde a pu être démontrée par les tests préopératoires (échocardiographie de stress à la dobutamine, IRM avec contraste, etc) [9].
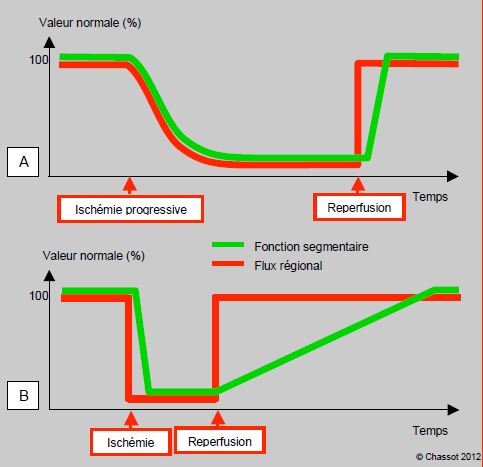
Figure 5.143 : Hibernation et sidération. A: hibernation; la fonction segmentaire suit la même évolution que la perfusion régionale. B : sidération ; la fonction, effondrée durant la période ischémique, ne remonte que lentement après le rétablissement de la perfusion.
L'examen échographique d'un coeur souffrant d'un infarctus frais laisse voir une zone dysfonctionnelle plus étendue que ne le sera la lésion cicatricielle ultérieure. C'est la zone bordante ou pénombre : la périphérie de la région ischémiée souffre d'une hypocontractilité associée à l'hypoperfusion, qui est une forme d’hibernation. Ce phénomène est réversible lors du rétablissement de la perfusion, alors que la partie centrale nécrosée ne peut plus récupérer.
Sidération (Stunning)
Ce phénomène consiste en une persistance de la dysfonction myocardique après revascularisation, alors que l'angor, le segment ST et la perfusion ont récupéré. Malgré le rétablissement de la perfusion coronarienne, la récupération n’est pas immédiate comme dans l'hibernation, et la fonction myocardique reste altérée pendant une période allant de quelques heures à plusieurs jours (Figure 5.143B) [4] ; la récupération peut parfois prendre plusieurs semaines. Cette lésion fonctionnelle survient à des degrés divers après angioplastie ou après pontage aorto-coronarien. Le myocarde lésé est caractérisé par des lésions ultrastructurales et électrophysiologiques sans nécrose, et par une perte de l'autorégulation coronarienne qui y rend le flux pression-dépendant. Il est "intoxiqué" par des radicaux libres (peroxydes) et par une augmentation du Ca2+ sarcoplasmique [9,23]. Bien que les réserves en ATP soient conservées, le couplage excitation-contraction est défaillant. La dysfonction est systolique et diastolique. Le myocarde sidéré est bien stimulable par des agents catécholaminergiques.
Lésions d’ischémie
Lorsque l’apport d’O2 est interrompu, la chaîne respiratoire mitochondriale cesse de fonctionner après une minute, la glycolyse passe en mode anaérobique et l’utilisation des acides gras est stoppée. Le pH intracellulaire commence à chuter et le taux de lactate à monter. En quinze minutes d'ischémie normothermique, l’ATP myocardique baisse de 65%. Vingt minutes suffisent à entraîner des foyers de nécrose myocardique irréversible; ceci survient lorsque le taux d’ATP est < 0.2 nmol/mg [3]. Bien qu’elle puisse fonctionner pendant une quarantaine de minutes, la glycolyse anaérobique ne peut que maintenir le fonctionnement des pompes ioniques, mais non assurer la contraction myocardique; le myocarde ne reste viable que s’il est arrêté. S'il est reperfusé à ce stade, il peut récupérer [8].
Lorsque les pompes dysfonctionnent à leur tour, le Na+ rentre massivement dans la cellule à cause de la chute d’activité des Na+/K+-ATPases et de l’échangeur Na+/H+ qui extrait les valences acides ; cette surcharge sodique provoque un œdème cellulaire. De son côté, le Ca2+ n’est plus repompé par le reticulum sarcoplasmique (RS) par manque d’ATP et s’accumule dans le cytoplasme, où il active les protéases, les lipases et les nucléases [3]. Les canaux MPTP de la paroi mitochondriale (mitochondrial permeability transition pore), qui sont normalement fermés, s’ouvrent lors d’une souffrance ischémique extrême. Cette ouverture signe l’arrêt de mort de la mitochondrie, car elle perd son imperméabilité : elle gonfle, son potentiel de membrane s’effondre, ses cristae se rompent, et ses composants propres comme le cytochrome c et les radicaux libres sont relargués dans la cellule, où ils activent les caspases (enzymes digérant les composants cellulaires) (Figure 5.144) [23]. Le cycle de Krebs s'arrête et la cellule meurt.
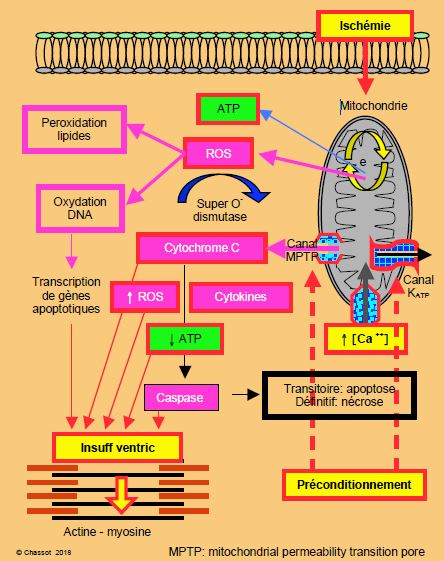
Figure 5.144 : Schématisation des lésions de l'ischémie. La mitochondrie laisse fuir (par le canal MPTP) des agents oxydants comme le cytochrome C, les ROS (Reactive Oxygen Species) ou superoxydes et des cytokines, qui vont freiner la synthèse d’ATP et activer les caspases qui digèrent la cellule (voir description détaillée sous Apoptose). e• : chaîne d’oxydo-réduction de la mitochondrie. MTPT : mitochondrial permeability transition pore. Le préconditionnement prévient partiellement l’apoptose en agissant sur deux points au niveau des mitochondries : 1) ouverture des canaux potassiques dépendants de l’ATP (KATP), ce qui diminue la charge en calcium ionisé (Ca2+) dans le cytosol mitochondrial, et 2) fermeture des canaux de perméabilité MPTP, ce qui diminue le taux de caspases cytoplasmiques d'après réf [22,23].
Lésions de reperfusion
Au lieu de rétablir la situation, la reperfusion du myocarde ischémié génère des lésions supplémentaires qui s’ajoutent à celles induites par l’interruption circulatoire, et met en route une cascade de phénomènes pouvant aboutir à des dégâts cellulaires irréversibles [3]. Elle cause un oedème explosif de la cellule et des organelles intracellulaires à cause du gradient osmotique massif attirant l'eau et les électrolytes (Na+, Ca2+) vers l'intérieur de la cellule. L'oedème cellulaire, la lésion des membranes et la libération d'enzymes cytotoxiques sont d'autant plus importants que le perfusat contient davantage d'oxygène et de calcium. Alors que l'hypothermie a un rôle protecteur pendant la phase ischémique, elle n'en a aucun pendant la phase de reperfusion. Toute une série de processus se met en route pendant cette période (Figure 5.145) [3,6,8,10,21].
- L’apport soudain d’O2 dans un milieu riche en radicaux libres va donner naissance à des produits très toxiques, les ROS (reactive oxygen species) ou radicaux libres (O2•, OH•, ONOO•); ceux-ci ne sont plus dégradés car l'ischémie a réduit les défenses naturelles contre ces superoxydes (superoxyde dismutase, réductases, etc).
- L'arrivée d'O2 dans une cellule démunie de ses défenses altère les membranes qui deviennent perméable au Ca2+.
- L’activation des canaux calcique lents (canaux L) et de l’échangeur Na+/Ca2+ qui extrait le Na+ fait rentrer une grande quantité de Ca2+ dans la cellule. L'hypercalcémie intracytoplasmique déjà présente à cause de l'ischémie est encore aggravée.
- La flambée de ROS ouvre les canaux MPTP de la paroi mitochondriale qui étaient restés fermés pour protéger la mitochondrie de l'acidose ischémique; les cations pénètrent ainsi dans la mitochondrie et découplent la chaîne d’oxydo-phosphorylation; la production d'ATP s'effondre. Si l'ouverture des MPTP concerne < 10% des mitochondries, la cellule peut récupérer, mais si > 50% des mitochondries sont atteintes, la nécrose survient.
- Les mitochondries utilisent l'oxygène pour le repompage du Ca2+ excédentaire et non pour la formation d'ATP. L'excès de Ca2+ y induit la formation de caspases, enzymes digérant les protéines cellulaires, qui fuient dans le cytoplasme par les canaux MTPT.
- La rigidification des myofibrilles (stone heart) due au blocage des ponts actine-myosine est secondaire à l’excès de Ca2+ libre dans le cytoplasme. Le ventricule est contracté et rigide, prompt à la fibrillation, et incapable de soutenir la circulation malgé des coronaires peméables.
- La production soudaine et excessive de NO• lève l’inhibition sur la transmigration leucocytaire et sur la régulation des superoxydes.
- La vasodilatation massive de la zone ischémiée, qui abolit l’autorégulation, est responsable d’un flux sanguin excessif malgré une pression de perfusion normale, et d’un risque d’œdème et/ou de surpression à l’intérieur de l’organe au moment de la reperfusion.
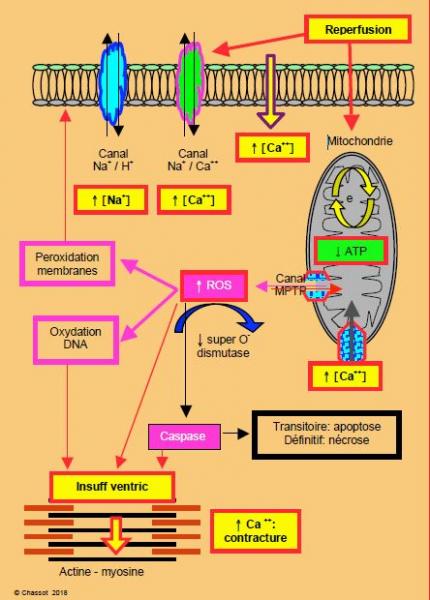
Figure 5.145 : Schématisation des lésions de reperfusion. La réactivation des canaux échangeurs Na+/H+ et Na+/Ca++ augmentent la concentration intracellulaire de Na+ et de Ca++. Les membranes laissent passer le Ca++. L'ouverture du canal MPTP (mitochondrial permeability transition pore) laisse entrer les cations dans la mitochondrie et sortir des agents oxydants comme le cytochrome C et les ROS (Reactive Oxygen Species), qui vont activer les caspases (digestion de la cellule), car les enzymes antioxydants font défaut après une période d'ischémie. e• : chaîne d’oxydo-réduction de la mitochondrie.
Il existe donc un paradoxe de l'oxygène et du calcium: alors que leur manque conduit à la nécrose ischémique, leur apport soudain à la cellule qui en a été privée déclenche une cascade d'évènements pathologiques qui sont plus graves que ne l’étaient les dégâts de l’ischémie. La reperfusion après un épisode ischémique est supposée guérir les lésions myocardiques ; or elle contribue à l’aggravation des dégâts cellulaires secondaires à l’occlusion vasculaire et compte pour moitié dans la taille finale de l’infarctus [8,10].
Des interventions thérapeutiques ciblées sur les mécanismes qui entraînent les dégâts au moment de la reperfusion ont un impact sur la taille des infarctus en expérimentation animale, mais n’ont malheureusement aucune application clinique efficace [8,21]. C’est notamment le cas des perfusions de glucose-insuline-potassium (GIK), de magnésium, d'anticalciques, d'antioxydants, de méthylprednisolone et d'autres substances actives sur les différents canaux transmembranaires (ranolazine, ciclosporine A, nicorandil), dont les résultats sont inconsistants [1,16]. D’autres possibilités de protection ont été envisagées sans plus de succès dans la pratique clinique, comme les peptides natriurétiques et les statines à haute dose qui agissent par l’activation des protéines-kinases accélérant la recapture de Ca2+ par le réticulum sarcoplasmique [14]. Pour l’instant, seuls le préconditionnement, le postconditionnement et le conditionnement à distance offrent une protection relative (voir Préconditionnement) [15].
Non-reperfusion (No-reflow)
Malgré la restauration d’un flux épicardique normal, le flux intramyocardique reste souvent compromis ou absent : c’est le phénomène de l'obstruction microvasculaire, ou no-reflow, qui augmente la taille de l’infarctus, la mortalité et les complications (arythmies, insuffisance ventriculaire). On estime que la reperfusion n’est totalement rétablie que dans 50-70% des cas d’angioplastie ou de revascularisation chirurgicale [17]. Les causes de la non-reperfusion sont multiples et probablement liées à une dysfonction endothéliale [3], mais les possibilités thérapeutiques sont très limitées [12,13,18].
- Lésions endothéliales capillaires, compression externe par l'œdème myocardique et l'hypercontracture due à la surcharge calcique de la reperfusion;
- Micro-embolisation distale d’athéromes, de thrombus et d’amas plaquettaires;
- Vasoconstriction distale intense (sécrétion réduite de NO, excès d'endothéline);
- Nécrose ischémique;
- Lésions de reperfusion;
- Conditions métaboliques : hyperglycémie, hypercholestérolémie.
Les arythmies
Les déséquilibres électro-chimiques liés à la reperfusion peuvent entraîner le dysfonctionnement électrique de certaines cellules et occasionner des arythmies ventriculaires malignes réfractaires aux thérapeutiques habituelles. Toutefois, ces arythmies sont en général réversibles si l’on parvient à maintenir la perfusion myocardique pendant quelques heures. Leur prise en charge demande beaucoup de persévérance : certains patients sont sortis de l’hôpital en rythme sinusal après avoir été défibrillés une cinquantaine de fois pendant les premières heures post-CEC. Lorsque le rythme est régulier, l'ECG montre des ondes T inversées, amples et pointues typiques des lésions de reperfusion.
| Ischémie et reperfusion |
|
Hibernation: forme d'autoprotection dans laquelle le myocarde ischémié devient hypocontractile pour adapter sa mVO2 au faible DO2. L'étendue est variable, mais il n'y a pas de nécrose. La fonction de ce myocarde est stimulable par des catécholamines; elle est récupérable en cas de revascularisation (en quelques heures à quelques jours).
Lésions de reperfusion: l’apport massif d’O2 et de calcium libère des superoxydes et des radicaux libres toxiques pour la cellule et s’accompagne d’une hypercalcémie sarcoplasmique. Dans un premier temps, la reperfusion aggrave l'état métabolique de la cellule myocardique. Bien que la pression soit normale, le flux sanguin est excessif par rapport à la vasodilatation maximale des zones ischémiées. La gravité des lésions de reperfusion est proportionnelle à celle des lésions ischémiques qui les précèdent et au contenu en O2 et en Ca2+ du sang qui reperfuse le myocarde. Des arythmies ventriculaires malignes sont fréquentes, mais potentiellement réversibles.
Sidération: persistance de la dysfonction myocardique après revascularisation alors que la perfusion est récupérée; la durée est de quelques heures à plusieurs jours; la fonction est stimulable par des catécholamines.
Non-reperfusion: absence de flux distal intra-myocardique alors que le flux épicardique est rétabli.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- ANTMAN E, COOPER H, McKINLEY S, et al. Early administration of intravenous magnesium to high-risk patients with acute myocardial infarction in the Magnesium in Coronaries (MAGIC) trial : a randomized controlled trial. Lancet 2002 ; 360 :1189-96
- ARAI AE, GRAUER SE, ANSELONE CG, et al. Metabolic adaptation to a gradual reduction in myocardial blood flow. Circulation 1995; 92:244.52
- BENHABBOUCHE S, CROLA DA SILVA C, ABRIAL M, FERRERRA R. The basis of ischemia-reperfusion and myocardial protection. Ann Fr Anesth Réanim 2011; 30:S2-S16
- BRAUNWALD E. Is myocardial stunning important from a clinical standpoint? In: Kloner RA, Przylenk K. ed. Stunned Myocardium. Marcel Dekker, Inc, 1993, p 441-452
- BRAUNWALD E, RUTHERFORD JD. Reversible ischemic left ventricular dysfunction: Evidence for the "hibernating myocardium". J Am Coll Cardiol 1986; 8:1467
- DE HERT SG, MOERMAN A. Myocardial injury and protection related to cardiopulmonary bypass. Best Pract Res Clin Anaesthesiol 2015; 29:137-49
- ELSASSER A, SCHLEPPER M, KLOVEKORN VP, et al. Hibernating myocardium: An incomplete adaptation to ischemia. Circulation 1997; 96:2920-31
- FERRARI R, BALLA C, MALAGU M, et al. Reperfusion damage – A story of success, failure, and hope. Circ Res 2017; 81:131-41
- FOËX P, BICCARD B. Stunning, hibernation, preconditioning, postconditioning, and the effects of anesthetic drugs. In: HOWELL S, et al, eds. Heart disease and the surgical patient. New York: Informa Healthcare 2007, 63-86
- FRÄSSDORF J, DE HERT S, SCHLACK W. Anaesthesia and myocardial ischaemia/reperfusion injury. Br J Anaesth 2009; 103:89-98
- HAAS F, JENNEN L, HEINZMANN U, et al. Ischemically compromised myocardium displays different time-courses of functional recovery: correlation with morphological alterations ? Eur J Cardiothorac Surg 2001; 20:290-8
- HAUSENLOY DJ, YELLON DM. Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol 2016; 13:193-209
- IBÁÑEZ B, HEUSCH G, OVIZE M, et al. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 2015; 65:1454-71
- KEVIN LG, NOVALIJA E, STOWE DF. Reactive oxygen species as mediators of cardiac injury and protection: the relevance to anesthesia practice. Anesth Analg 2005; 101:1275-87
- KLONER RA, BROWN DA, CSETE M, et al. New and revisited approaches to preserving the reperfused myocardium. Nat Rev Cardiol 2017; 14:679-93
- MEHTA SR, YUSUF S DIAZ R, et al. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction : the CREATE-ECLA randomized controlled trial. JAMA 2005 ; 293 :437-46
- NICCOLI G, BURZOTTA F, GALIUTO L. Myocardial no-reflow in humans. J Am Coll Cardiol 2009; 54:281-92
- PRASAD A, STONE GW, HOLMES DR, GERSH B. Reperfusion injury, microvascular dysfunction, and cardioprotection. Circulation 2009; 120:2105-12
- RAHIMTOOLA SH. The hibernating myocardium. Am Heart J 1989; 117:211-21
- SHAN R, BICK RJ, POINTDEXTER DJ, et al. Altered adrenergic receptor density in myocardial hibernation in humans: A possible mechanism of depressed myocardial function. Circulation 2000; 102:2599-606
- YELLON DM, HAUSENLOY DJ. Myocardial reperfusion injury. N Engl J Med 2007; 357:1121-35
- ZAUGG M, LUCHINETTI E, UECKER M, PASCH T, SCHAUB MC. Anaesthetics and cardiac preconditioning: signaling and cytoprotective mechanisms. Part I. Brit J Anaesth 2003 ; 91:551-65
- ZAUGG M, SCHAUB MC, FOËX P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21-33