Il est capital que la réaction coagulatoire ne s’emballe pas. Elle doit rester localisée à l’endroit de la lésion vasculaire afin de stopper l’hémorragie, mais ne doit en aucun cas provoquer des thromboses intempestives disséminées dans tout l’arbre vasculaire. Plusieurs mécanismes permettent ce contrôle (Figure 8.7).
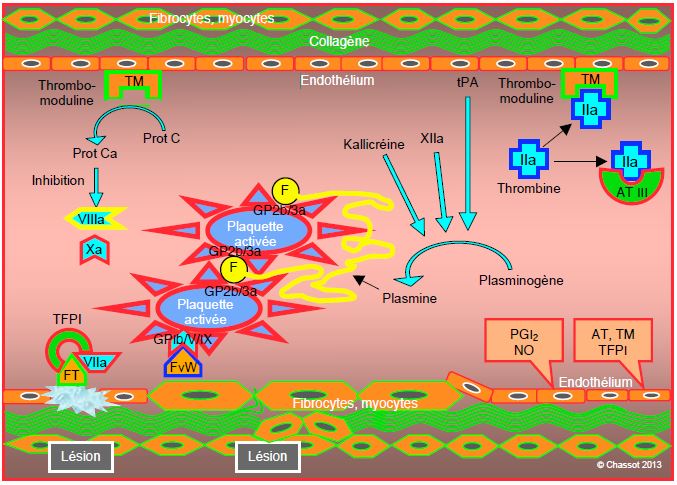
Figure 8.7: Limitation. Plusieurs systèmes empêchent la diffusion de la coagulation: 1) la thrombomoduline (TM) endothéliale inhibe la thrombine circulante; 2) le complexe TM-thrombine active la protéine C (Prot Ca) qui bloque les facteurs Va et VIIIa; 3) l’antithrombine III (AT III) inactive rapidement la thrombine et plus lentement le facteur IXa; 4) la prolifération du complexe FT/VIIa est limitée par son inhibiteur circulant (TFPI, tissue factor pathway inhibitor); 5) la fibrinolyse assure la dégradation progressive des amas de fibrine et la résorbtion du thrombus; elle est assurée par la plasmine issue du plasminogène sous l’action de la kallikréine, du facteur XIIa et de l’activateur endothélial (tPA, tissue plasmin activator). PG: prostaglandine.
- La fibrinolyse;
- Les inhibiteurs circulants;
- L’activité endocrine des plaquettes;
- L’endothélium vasculaire sain;
- La contrainte pour la chaîne coagulatoire de se dérouler à la surface de plaquettes activées par la lésion endothéliale.
Fibrinolyse
Une fois l’hémostase réalisée et la paroi vasculaire étanche, le thrombus doit disparaître. La fibrinolyse consiste en un clivage de la fibrine en fragments sans activité coagulatoire (D-dimères). Elle est réalisée par la plasmine, transformée à partir du plasminogène circulant par deux déclencheurs principaux [1].
- L’activateur tissulaire du plasminogène (t-PA, tissue plasminogen activator), synthétisé dans l’endothélium vasculaire en réponse au thrombus et à la stase;
- L’activateur sérique du plasminogène, ou urokinase (u-PA, urokinase plasminogen activator), activé à partir de la pro-urokinase par la plasmine, le facteur XIIa et la kallikréine ; il se rencontre en plus grande quantité dans le système urinaire.
La fibrinolyse est également contrôlée par deux systèmes freinateurs: des inhibiteurs de l’activation du plasminogène (plasminogen activator inhibitors PAI-1 et PAI-2), et l’inhibiteur de la fibrinolyse activé par la thrombine (TAFI, thrombin-activated fibrinolysis inhibitor) qui réduit les points d’attache pour la plasmine.
La fibrinolyse peut elle aussi déraper et devenir excessive, au point que la plasmine dégrade également le fibrinogène. Elle est donc régulée par un inhibiteur de ses activateurs spécifiques. Dans les cas cliniques, elle peut être freinée par des agents antifibrinolytiques comme l’aprotinine, l’acide tranexamique ou l’acide ε-aminocaproïque. L’acide tranexamique et l’acide ε-aminocaproïque se fixent sur la lysine du plasminogène et bloquent l'activation de la plasmine. L’aprotinine est un inhibiteur non spécifique des protéases, qui bloque directement la plasmine. En chirurgie cardiaque, ces substances diminuent globalement les pertes sanguines de 30% et les reprises chirurgicales pour hémorragie de 60% [2,6].
Inhibiteurs
Comme on l’a déjà mentionné, plusieurs mécanismes associés à la cascade coagulatoire jouent un rôle capital pour limiter l’extension du caillot. Lorsqu’ils s’échappent dans la circulation, les facteurs de coagulation sont ainsi rapidement maîtrisés [1,5,7].
- La prolifération du complexe FT/VIIa en-dehors de la lésion endothéliale est limitée par son inhibiteur circulant (TFPI, tissue factor pathway inhibitor), essentiellement produit par l’endothélium; il inhibe rapidement tout complexe s’échappant en-dehors de la zone lésée.
- La thrombomoduline (TM) endothéliale inhibe la thrombine.
- Le complexe TM-thrombine active la protéine C et la protéine S qui bloquent les facteurs Va et VIIIa.
- L’antithrombine III (AT III) inactive rapidement la thrombine et le facteur Xa, plus lentement les facteurs IXa, XIa et XIIa; elle est responsable de plus de la moitié de l’effet inhibiteur total. L’héparine agit comme un cofacteur le l’AT III et rend la réaction inhibitrice de la thrombine 2’000 fois plus active.
- La thrombine en excès est liée à la fibrine qui vient de se former (antithrombine I).
- Le facteur VIIIa est instable et se dissocie spontanément.
Endothélium
L’endothélium est une couche cellulaire extrêmement active qui possède un rôle crucial dans la protection contre les thromboses. L’endothélium sécrète des substances qui freinent la coagulation et modifient la perfusion locale.
- Inhibiteur du complexe FT/VIIa (TFPI, tissue factor pathway inhibitor).
- Activateur tissulaire de la plasmine (tPA): activation de la fibrinolyse.
- Thrombomoduline : inhibition des facteurs IIa (thrombine) et Xa.
- Récepteur de la protéine C : inhibition des facteurs Va et VIIIa.
- NO•: principal agent vasodilatateur distal (baisse du Ca2+ ionisé intracellulaire), inhibition de l’agrégation plaquettaire.
- Prostacycline PGI2: augmentation du cAMP, diminution de l’agrégation plaquettaire.
- Ecto-ADPase: suppression de la phase de recrutement plaquettaire.
- Endothéline: vasconstricteur puissant, dont la sécrétion est stimulée par la thrombine, l'angiotensine II, l'adrénaline et la vasopressine.
Un endothélium malade réagit de manière pathologique. Lors de réaction inflammatoire majeure ou d’athéromatose, par exemple, la balance physiologique entre les effets du NO• et ceux de l’endothéline sont modifiés au profit d'une vasocontriction et d’une stimulation plaquettaire [9]. Ainsi, des stimuli qui entraînent normalement une vasodilatation (hypoxie, exercice et augmentation de la demande en O2, par exemple) peuvent conduire à une vascoconstriction et à une augmentation de l’adhésivité plaquettaire dans des vaisseaux artériosclérotiques. Dans certaines situations anormales, l’endothélium peut exprimer le facteur tissulaire (FT), et de ce fait devenir un déclencheur de la coagulation. C’est le cas dans l’exposition à certains lipopolysaccharides bactériens (sepsis), aux cytokines inflammatoires ou au LDL oxydé [1]. Par ailleurs, le FT est un composant majeur des plaques athéromateuses [8].
Plaquettes
Lorsqu’elles sont stimulées, les plaquettes libèrent des agents activateurs de la coagulation (TXA2, ADP, sérotonine, thrombine) dont certains sont aussi des vasoconstricteurs locaux (TXA2, sérotonine), alors que l’endothélium sécrète des substances qui freinent l’activité plaquettaire et ont un effet vasodilatateur : NO•, prostacycline PGI2 et ecto-ADPase. La situation peut évoluer de deux manières différentes [4].
- Prédominance des agents vasodilatateurs et inhibiteurs des plaquettes : résolution spontanée du thrombus ; la circulation est rétablie, mais la cicatrisation et la fibrose de la plaque athéromateuse accroissent progressivement sa taille.
- Prédominance des agents vasoconstricteurs et stimulateurs des plaquettes comme le stress, la fumée ou l’inflammation : recrutement massif de thrombocytes et de facteurs de coagulation ; le thrombus devient occlusif et provoque une ischémie ou une nécrose distale.
L’activité endocrine opposée des plaquettes et de l’endothélium maintient un équilibre dynamique au niveau artériolaire. Mais certaines situations sont accompagnées d’une excitabilité plaquettaire exagérée : obésité, tabagisme, hyperlipidémie, hypercholestérolémie, hypertension artérielle, vieillissement, diabète, insuffisance rénale [3]. La balance penche alors en faveur de la thrombose.
| Systèmes régulateurs de la coagulation |
|
Plusieurs mécanismes permettent à la coagulation de rester cantonnée à l’endroit de la lésion vasculaire sans risque de dissémination :
- Fibrinolyse : clivage de la fibrine par la plasmine
- Inhibiteurs circulants : anti-thrombine (AT III), thrombomoduline (inhibiteur de thrombine)
inhibiteur du complexe FT/VIIa
- Dissociation spontanée : facteur VIIIa
- Endothélium (action inhibitrice) : inhibition du complexe FT/VIIa, activation de plasmine,
sécrétion de NO, de prostacycline PGI2, d’endothéline
- Présence obligatoire de plaquettes : seule des plaquettes activées et localement fixées
offrent une surface adéquate pour le déroulement de la cascade coagulatoire
|
© CHASSOT PG, MARCUCCI C, Décembre 2013, dernière mise à jour, Novembre 2018
Références
- ADAMS RLC, BIRD RJ. Review article: Coagulation cascade and therapeutic update: Relevance to nephrology. Part I: Overview of coagulation, thrombophilia and history of anticoagulants. Nephrol 2009; 14:462-70
- BROWN JR, BIRKMEYER NJO, O’CONNOR GT. Meta-analysis comparing the effectiveness and adverse outcomes of antifibrinolytic agents in cardiac surgery. Circulation 2007; 125:2801-13
- DAVI G, PATRONO C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482-94
- FALK E, SHAH PK, FUSTER V. Coronary plaque disruption. Circulation 1995; 92:657-71
- INNERHOFER P, KIENAST J. Principles of perioperative coagulopathy. Best Pract Res Clin Anaesthesiol 2010; 24:1-14
- KOSTER A, SCHIRMER U. Re-evaluation of the role of antifibrinolytic therapy with lysine analogs during cardiac surgery in the post aprotinin era. Curr Opin Anaesthesiol 2011; 24:92-7
- MOSESSON MW. Update on antithrombin I (fibrin). Thromb Haemost 2007; 98:105-8
- VILES-GONZALEZ JF, ANNAND SX, ZAFAR MU, et al. Tissue factor coagulation pathway: A new therapeutic target in atherothrombosis. J Cardiovasc Pharmacol 2004; 43:669-76
- ZAUGG M, SCHAUB MC, FOËX P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21-33