Au niveau cellulaire, l’insuffisance ventriculaire se caractérise par de nombreux dysfonctionnements, dont les quatre principaux sont directement liés aux processus précédemment décrits (Figure 5.8).
- La recapture du Ca2+ par le RS est diminuée à cause d'une suractivité du phospholamban, qui inhibe l’activité de la SERCA [5,10,12,15]. Les réserves de Ca2+ du RS sont insuffisantes pour assurer un relargage adéquat lors de la systole (faible calcium-induced calcium release). Les récepteurs R-Ry laissent fuir du Ca2+ en diastole, ce qui surcharge de cytoplasme [14].
- L’activité de la β-ARK est excessive à cause de la stimulation sympathique β permanente. Le récepteur β est découplé de la protéine Gs, donc la chaîne qui mène à la synthèse d'AMPc devient insuffisante. L’activité de la protéine Gi (inhibitrice) est prépondérante [11].
- Sur les récepteurs β, la protéine G inhibitrice a un effet prédominant ; de ce fait, la quantité d’AMPc synthétisé est moindre, ce qui conduit à une plus faible quantité de Ca2+ libéré dans le sarcoplasme lors de chaque stimulation. Les variations de la concentration de Ca2+ sont moins amples, et la force de contraction des myofibrilles diminue.
- La sur-stimulation sympathique chronique entraîne une désensibilisation et une diminution du nombre des récepteurs β1 (down-regulation). Ils représentent moins de 40% du total des récepteurs (au lieu de 80%, une diminution de moitié) [17]. La proportion des récepteurs β2 (chronotropes positifs) augmente à 40% et celle des récepteurs β1 (inotropes positifs) à 20% ; les récepteurs β3, inotropes négatifs couplés au NO•, sont beaucoup plus nombreux [1,3,4,6,18].
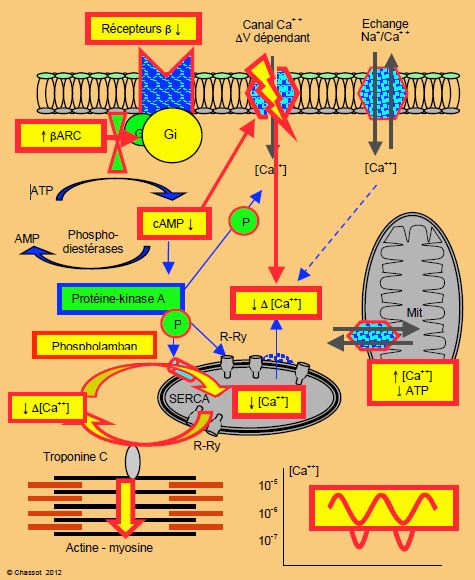
Figure 5.8 : Schéma des modifications du couplage excitation – contraction en cas d’insuffisance ventriculaire. Les variations de la ([Ca++]i) sont diminuées par une série de processus (voir description détaillée dans le texte).
Ces phénomènes conduisent à une diminution de la réserve en Ca2+ du RS et à une diminution de l’amplitude des variations de la [Ca2+]i. La force de contraction en est affaiblie d’autant (insuffisance systolique). La stagnation du Ca2+ dans le sarcoplasme conduit à une insuffisance diastolique et à une accumulation de Ca2+ dans les mitochondries, ce qui abaisse la production d’ATP [13]. Le cœur défaillant retourne à une expression génique de type fœtal, dont un marqueur est la production excessive de Brain natriuretic peptide (BNP) par le ventricule. Celui-ci perd de son efficacité énergétique en mobilisant trop d’acides gras pour son métabolisme à cause de la sur-stimulation sympathique [13,22]. Le taux de noradrénaline dans le sinus coronaire de cœurs en insuffisance congestive est de 50 fois la norme, ce qui indique bien que le myocarde ne répond plus correctement à la stimulation sympathique [21]. Le NO• a une relation biphasique avec la fonction myocardique : une faible et constante production de NO• est bénéfique au fonctionnement cellulaire systolique et diastolique ; elle a une action cardioprotectrice et vasodilatatrice. Une production élevée, au contraire, inhibe la respiration mitochondriale, freine la contractilité, bloque l’activité des amines β et induit une vasoplégie massive [7,10].
| Dysfonction ventriculaire |
|
Au niveau cellulaire, l'insuffisance ventriculaire se caractérise par:
- Activité excessive du phospholamban: inhibition des mouvements du Ca2+ du réticulum sarcoplasmique
- Activité excessive de la β–ARK: inhibition des récepteurs β
- Hyperactivité de la protéine inhibitrice Gi et frein de la synthèse d'AMPc (baisse de Ca2+)
- Désensibilisation et baisse du taux de récepteurs β (downregulation)
La concentration myocardique des récepteurs β1 baisse de moitié. La compensation est une augmentation du taux de récpeteurs β2 (chronotropes et inotropes positifs) et β (inotropes positifs).
|
Traitement de l’insuffisance ventriculaire: les β-bloqueurs
Les β-bloqueurs font très logiquement partie de la première ligne de traitement de l’insuffisance cardiaque parce qu’ils contrecarrent plusieurs dysrégulations pathognomoniques de la dysfonction du ventricule (Figure 5.9) [3,8,16,20].
- Diminution de l’activité de la β-ARK ; cet effet rétablit la sensibilité des récepteurs β aux catécholamines endogènes.
- Maintien du nombre des récepteurs β par compétition avec l’excès d’amines circulantes qui induisent une baisse des β1 myocardiques.
- Augmentation de l’activité de la SERCA, donc accélération de la recapture du Ca2+ par le RS, et diminution de la fuite diastolique de Ca2+ par les canaux R-Ry. La contraction et la fonction diastolique sont améliorées par baisse du taux résiduel de Ca2+ dans le cytoplasme et augmentation du Ca2+ à disposition dans le RS pour la libération systolique.
- Déplacement du métabolisme mitochondrial vers une utilisation du glucose plutôt que des acides gras, ce qui augmente le taux d’ATP produit par rapport à la quantité de substrat utilisée.
- Augmentation de la sécrétion d’ANF (Atrial natriuretic factor) et de BNP (voir Modifications neuro-endocrines).
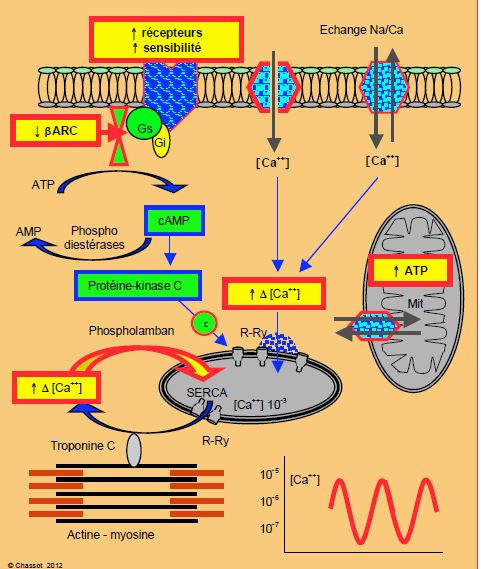
Figure 5.9 : Schématisation de l’action des β-bloqueurs dans l’insuffisance cardiaque pour rétablir une augmentation des variations de la [Ca++]i.
Traitement de l’insuffisance cardiaque : les substances inotropes
Les agents inotropes positifs agissent par la voie commune d’une augmentation de la libération du Ca2+ par le RS lors de la systole (Figure 5.10) [2,9].
- Les catécholamines agissent par le biais d’une augmentation de l'AMPc en stimulant l’adénylate-cyclase; en cas d'inhibition des récepteurs β1, seules les amines à effet β2 et alpha restent pleinement efficaces (adrénaline).
- Les anti-phospho-diestérases-3 (amrinone, milrinone) inhibent la dégradation de l'AMPc. Agissant par une autre voie que les amines β, ces substances restent efficaces même en cas de β-blocage profond ou de régulation à la baisse des récepteurs β.
- La combinaison adrénaline – milrinone est particulièrement indiquée en cas d'insuffisance ventriculaire chronique avec dowregulation des récepteurs β.
- La digitale intervient sur les canaux échangeurs Na+/ Ca2+ de la membrane cellulaire en favorisant l’entrée de Ca2+.
- Le levosimendan fonctionne comme un sensibilisateur calcique; il interagit directement avec la troponine C et stabilise sa liaison avec le Ca2+, ce qui amplifie et prolonge la contraction [19]. Cet effet dépend de la [Ca2+]i, mais non de l'état du système β-adrénergique. De plus, le levosimendan agit sur les canaux KATP membranaires (canaux potassiques ATP-dépendants) qui, dans les cellules musculaires lisses des artères, provoquent une relaxation et une vasodilatation.
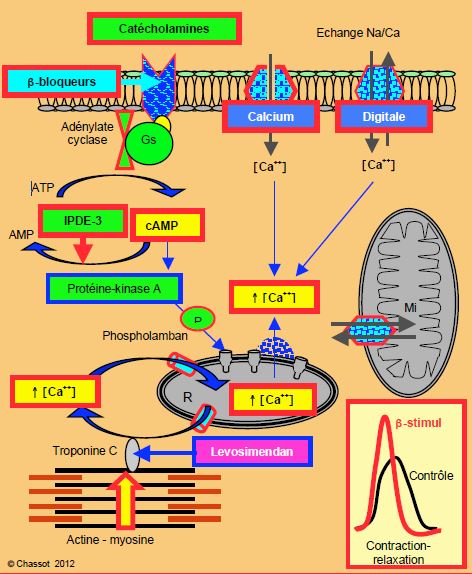
Figure 5.10 : Schématisation des points d’impact des différents agents inotropes positifs. A l’exception du levosimendan qui agit au niveau de la fixation du Ca++ sur la troponine C, tous aboutissent à une augmentation des variations de la [Ca++]i. IPDE-3 : inhibiteur des phosphodiestérases-3 (amrinone, milrinone). Les β-bloqueurs sont mentionnées ici à cause de leur impact dans le traitement de l’insuffisance ventriculaire chronique.
| Implications pour le traitement de la dysfonction ventriculaire |
|
L'efficacité des amines β1 (dobutamine, dopamine) est faible à cause de la diminution des récepteurs β1 dans la défaillance ventriculaire. L'adrénaline (stimulant β1, β2 et α) reste efficace, de même que les substances qui agissent par une autre voie: antiphosphodiestérases-3 (milrinone), sensibilisateur calcique (levosimendan).
Les β-bloqueurs inhibent les systèmes de régulation à la baisse des récepteurs β et maintiennent une stimulation moycardique adéquate.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- BOHM M. Catecholamines refractoriness and their mechanisms in cardiocirculatory shock and chronic heart failure. J Thorac Cardiovasc Surg 1998; 46(suppl 2):270-5
- BRAUNWALD E. Heart failure. J Am Coll Cardiol HF 2013; 1:1-20
- BRISTOW MR. Antiadrenergic therapy of chronic heart failure. Surprises and new opportunities. Circulation 2003; 107:1100-2
- BRISTOW MR, GINSBURG R, MINOBE W, et al. Decreased catecholamine sensitivity and beta-adrenergic receptor density in failing human hearts. N Engl J Med 1982; 307:205-11
- BRITTSAN AG, KRANIAS EG. Phospholamban and cardiac contractile function. J Mol Cell Cardiol 2000; 32:2131-7
- BRODDE OE, DAUL A, MICHEL-REHNER L, et al. Agonist-induced desensitization of beta-adrenoreceptor function in humans. Circulation 1990; 81:914-21
- DREXLER H. Nitric oxide synthase in the failing heart: a double-edged sword ? Circulation 1999; 99:2971-5
- FOODY JM, FARREL MH, KRUMHOLZ HM. -blocker therapy in heart failure. Scientific review. JAMA 2002; 287:883-9
- GROBAN L, BUTTERWORTH J. Perioperative management of chronic heart failure. Anesth Analg 2006; 103:557-75
- HARTUPEE J, MANN DL. Neurohumoral activation in heart failure with reduced ejection fraction. Nat Rev Cardiol 2017; 14:30-8
- IWASE M, BISHOP SP, UECHI M, et al. Adverse effects of chronic endogenous sympathetic drive induced by cardiac Gsa overexpression. Circ Res 1996; 78:517-24
- KOSS KL, KRANIAS EG. Phospholamban: a prominent regulator of myocardial contractility. Circ Res 1996; 79:1059-63
- MAGNER JJ, ROYSTON D. Heart Failure. Br J Anaesth 2004; 93:74-85
- MARKS AR, REIKEN S, MARX SO. Progression of heart failure. Is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor ? Circulation 2002; 105:272-5
- MÜNCH G, et al. SERCA 2a activity correlates with force-frequency relationship in human myocardium. Am J Physiol 2000; 278:H1924-H1932
- PACKER M, COATS AJS, FOWLER MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med 2001; 344:1651-8
- PORT JD, BRISTOW MR. Altered beta-adrenergic receptor gene regulation and signalling in chronic heart failure. J Mol Cell Cardiol 2001; 33:887-905
- SHAN R, BICK RJ, POINTDEXTER DJ, et al. Altered adrenergic receptor density in myocardial hibernation in humans: A possible mechanism of depressed myocardial function. Circulation 2000; 102:2599-606
- TOLLER WG, STRANZ C. Levosimendan, a new inotropic and vasodilator agent. Anesthesiology 2006; 104:556-69
- YEAGER MP, FILLINGER MP, HETTLEMAN BD, et al. Perioperative beta-blockade and late cardiact outcomes: A complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237-41
- ZAUGG M, SCHAUB MC. Cellular mechanisms in sympatho-modulation of the heart. Br J Anaesth 2004; 93:34-52
- ZAUGG M, SCHAUB MC, FOËX P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21-33