La valeur de pression artérielle maximale considérée comme normale chez un adulte est 130/80 mmHg. Au-delà commence le domaine de la pré-hypertension (jusqu’à 140/90 mmHg) et de l’hypertension (≥ 140/90 mmHg) [19]. Ces valeurs peuvent paraître basses, mais la préhypertension double le risque cardiovasculaire à 10 ans [18]. Le meilleur prédicteur du risque cardio-vasculaire est la pression pulsée, ou PA différentielle (PAsyst – PAdiast). Sa valeur normale est 50-60 mmHg ; le risque d’accident cardio-vasculaire augmente significativement lorsqu’elle est > 80 mmHg [12]. Cela tient au fait que dans le cœur, le cerveau et les reins, les RAS locales sont basses ; la pulsatilité artérielle s’y propage très distalement, alors que le flux est quasi-dépulsé au-delà des petites artérioles dans les muscles et les viscères. Si la pulsatilité augmente, ces organes souffrent tout particulièrement et réagissent en augmentant l’épaisseur de paroi de leurs vaisseaux, mais le stress subi à chaque cycle cardiaque augmente le risque d’accident vasculaire (infarctus, ictus, néphropathie).
Le moteur ventriculaire n’est pas le seul élément à intervenir dans le contrôle de la pression artérielle. Il existe encore une série d’éléments qui se manifestent à différents niveaux.
Baroréflexes
Les fibres efférentes du sinus carotidien et des barorécepteurs de l’arc aortique cheminent par le nerf glossopharyngien (IX) jusqu'au ganglion pétreux, et de là à l'hypothalamus (noyau solitaire). Elles sont stimulées par une distension de la paroi vasculaire et par la pulsatilité du flux au niveau du récepteur. Par réflexe, ceci conduit à une stimulation vagale (via le nerf vague, X) qui induit rapidement une bradycardie et une vasodilatation artériolaire (Figure 5.71). Le délai de la boucle réflexe est bref: 1 à 2 secondes [8]. A l'inverse, une hypotension ou une dépulsation artérielle au niveau du sinus carotidien fait cesser l'émission de ce dernier, ce qui lève le frein parasympathique et provoque une augmentation du tonus sympathique (tachycardie, vasoconstriction artériolaire, veinoconstriction centrale). Cela survient en cas d'hypotension ou d'hypovolémie.
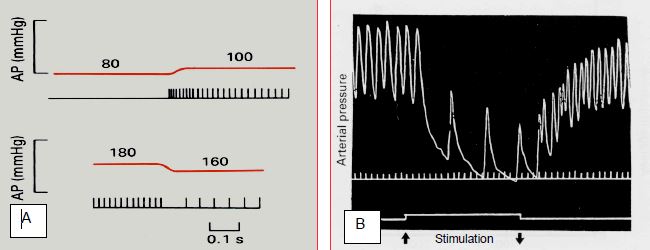
Figure 5.71 : Enregistrements de la pression artérielle illustrant le baroréflexe. A: Emissions d'un sinus carotidien de chat lorsque la pression augmente ou diminue; la fréquence des signaux émis est maximale lors de la montée de pression; elle diminue lorsque la pression est stable. Une chute de pression inhibe momentanément les émissions du sinus. B: Effet sur la pression artérielle et la fréquence cardiaque d'une stimulation électrique du sinus carotidien chez le chien (entre les deux flèches); les intervalles de temps représentent 0.2 seconde [Extrait de: Levick JR. An introduction to cardiovascular physiology. 2nd edition. Oxford: Butterworth-Heinemann, 1995, Figures 14.3 et 14.5].
Le baroréflexe est particulièrement sensible aux variations de pression; son émission est maximale pendant l'ascension de la valeur de pression artérielle. Lorsque la pression se stabilise, l'intensité de l'émission diminue. Après une quinzaine de minutes, le système se règle sur cette nouvelle valeur et la maintient (resetting). Le baroréflexe est donc efficace pour la régulation et le maintien de la pression à court terme [3]. Le baroréflexe est altéré ou supprimé dans les lésions du sinus carotidien (irradiation du cou, paragangliome), dans la dysautonomie familiale, après chirurgie carotidienne, ou dans certaines maladies neurodégénérative (Parkinson). La symptomatologie est caractérisée par une extrême labilité tensionnelle; elle consiste en crises hypertensives imprévisibles, le plus souvent déclenchées par le stress, en épisodes hypotensifs et en hypotension orthostatique. Les variations de pression ne s'accompagnent pas de modification réciproque de la fréquence cardiaque. La normalisation de la pression est quasi-impossible, malgré la prescription de méthyldopa, de guanfacine, de bloqueur mixte alpha-béta, de diazépine ou de dexmédétomidine trans-nasale [1].
Le glomus carotidien se situe dans le voisinage du sinus, à l'origine de la carotide interne. Ses cellules sont sensibles à la pO2. Il est stimulé par une hypoxie locale; via le nerf glosso-pharyngien, ceci entraîne une stimulation sympathique et une hypertension. L'effet hémodynamique est donc l'inverse de celui d'une stimulation du sinus carotidien [20].
Du côté veineux, des récepteurs situés dans les oreillettes et dans l’artère pulmonaire sont sensibles à la tension de la paroi. Ils sont stimulés par l’hypervolémie, envoient des signaux par le nerf vague jusqu’à l’hypothalamus et inhibent la stimulation sympathique et la sécrétion de rénine. Ils contrôlent le remplissage veineux. Ils sécrètent le facteur natriurétique auriculaire (Atrial natriuretic peptide ou ANF), qui a un effet diurétique, natriurétique et bradycardisant. La libération de BNP (Brain natriretic peptide) par le ventricule est aussi l’effet de la tension de paroi, mais cette fois-ci dans les myocytes ventriculaires. Le BNP provoque une diurèse et une natriurèse, et possède également des effets vasodilatateurs artériels et veineux (baisse de précharge et de postcharge) ; il inhibe la sécrétion de rénine [7]. Les taux circulants de BNP sont directement proportionnels au degré de surcharge de la paroi ventriculaire, ce qui explique leur valeur prédictive pour le diagnostic de l’insuffisance ventriculaire [16].
D’autres récepteurs cardiaques sont sensibles à l’hypovolémie. Une brusque chute dans la tension de la paroi ventriculaire sur une baisse de précharge déclenche le réflexe de Bezold-Jarisch qui associe une bradycardie vagale, une hypotension et une vasodilatation artérielle ; au niveau cardiaque, il est accompagné d’une vasodilatation coronarienne et d’un effet inotrope négatif [21]. La finalité de ce réflexe vagal est de maintenir le remplissage ventriculaire en ralentissant la fréquence. Normalement, il s’associe aux autres baroréflexes pour contrôler la pression et le débit artériel [6]. Mais il peut dominer la régulation lorsque le retour veineux est diminué de manière soudaine, comme lors de l’appui d’un utérus gravide sur la veine cave inférieure chez la parturiente en décubitus dorsal. Il peut aussi se déclencher en cas d’hypovolémie lorsque le retour veineux est compromis par une surpression abdominale ; le bas débit associé à l’hypotension est alors potentialisé par la bradycardie. Comme les récepteurs cardio-inhibiteurs sont les plus nombreux dans la paroi inféro-postérieure du VG, il n’est pas rare de voir une bradycardie et une hypotension accompagner un infarctus dans cette localisation [14]. Ce réflexe participe à la bradycardie qui peut se déclencher lors de bloc spinal, mais n’est pas seul en cause dans ce contexte [6]. Il est en effet associé au réflexe de Bainbridge et au réflexe "inversé" de Bainbridge. Le réflexe de Bainbridge consiste en une tachycardie associée à une augmentation du remplissage vasculaire central, expliquée par une suppression du tonus vagal. Cette régulation fait varier la fréquence de 20% chez l’homme, mais de 100% chez le chien [2]. Il est à l’origine de la l’augmentation de fréquence cardiaque associée à l’inspirium. Le réflexe "inversé" de Bainbridge est caractérisé par une bradycardie engendrée par la baisse du retour veineux et va dans le même sens que le réflexe de Bezold-Jarish, mais il est déclenché par les récepteurs au volume situés dans les veines centrales et l’OD, alors que le second est déclenché par les récepteurs situés principalement dans le VG [2].
Contrôle neuro-humoral de la pression
L’angiotensine II (AT-II) est issue de la transformation en deux étapes de l'angiotensinogène: la rénine sécrétée par l’appareil juxtaglomérulaire rénal le transforme en angiotensine I et l'enzyme de conversion en angiotensine II. La rénine répond à trois stimuli : une stimulation β- adrénergique, une chute de pression dans l’artère rénale et une fuite tubulaire de sodium. L’AT-II provoque une série d’effets [17].
- Vasoconstriction artérielle par effet direct sur les cellules musculaires lisses, par facilitation de la sécrétion de nor-adrénaline dans les fibres sympathiques efférentes et par stimulation de la production d’endothéline par l’endothélium vasculaire;
- Régulation des baroréflexes à un niveau de pression supérieur;
- Vasoconstriction de l’artériole efférente des glomérules (augmentation de la filtration glomérulaire);
- Rétention tubulaire de sodium;
- Stimulation de la sécrétion d’aldostérone par la corticosurrénale (résorbtion de sodium et d’eau) et d’ADH par la posthypophyse (rétention d’eau);
- Activation de la croissance et de l’hypertrophie myocardique;
- Activation de la fibrose et de l'apoptose;
- Libération de radicaux libres, inhibition de la sécrétion de NO, effet inflammatoire.
L'activité de l'AT-II est produite par l'activation des récepteurs membranaires AT1 essentiellement. L'activation des récepteurs AT2 provient principalement de l'angiotensine 1-7 (AT1-7), issue de l'AT-II, qui a des effets inverses: vasodilatation, production de NO, effet anti-inflammatoire, inhibition de la croissance et de la fibrose [4]. Le système rénine-angiotensine possède donc sont propre mécanisme d'autorégulation.
La dysautonomie est caractérisée par une mauvaise régulation de la pression artérielle, se présentant sous forme d’hypotension orthostatique (baisse de > 20/10 mmHg à la station verticale). Elle est due à un niveau de nor-adrénaline anormalement bas. De ce fait, les récepteurs adrénergiques vasculaires sont en grand nombre (upregulation), ce qui entraîne une hypersensibilité aux agents vasopresseurs [11]. Outre ses formes familiale et idiosyncrasique, la dysautomie se rencontre dans une série d’affections : diabète, sénescence, maladie de Parkinson, lésion médullaire, amyloïdose.
Contrôle périphérique des résistances
La variation du diamètre des petits vaisseaux est une manière très efficace de modifier les résistances (RAS), puisque celles-ci varient avec la quatrième puissance du rayon d’un tube (loi de Poiseuille). Plusieurs systèmes sont en cause pour régler localement les RAS.
- Récepteurs vasoconstricteurs : α1-adrénergiques, récepteurs à l’angiotensine (AT-II), endothéline.
- Système vasodilatateur du cGMP de la cellule musculaire lisse: récepteurs β2-adrénergiques, NO•.
- Facteurs endothéliaux : production de NO• (vasodilatateur) et d’endothéline (vasoconstricteur).
- Autres vasodilatateurs : prostacycline, bradykinine, histamine, adénosine, ions H+ liés à l’activité métabolique locale.
Rôle de l’endothélium
Longtemps considéré comme un simple revêtement anti-thrombogène, l’endothélium est au centre d’une cascade de régulations métaboliques, hémostatiques, immunitaires, inflammatoires et hémodynamiques. Il participe à la régulation des résistances artérielles par plusieurs mécanismes [5,9,13].
- NO• : synthétisé à partir de la L-arginine par la NO-synthase en fonction de l’augmentation du flux sanguin, de sa pulsatilité et des forces de cisaillement exercées sur l’endothélium, il est un puissant vasodilatateur local qui ajuste le diamètre du vaisseau au débit. Il freine l’adhésivité plaquettaire et la réponse inflammatoire.
- Endothéline : puissant vasoconstricteur par stimulation des récepteurs ETA de la musculature lisse, elle induit aussi une hyperplasie cellulaire (cellules musculaires lisses et fibroblastes).
- Prostacycline : produite en réponse à une stimulation inflammatoire (TNFα, IL-1), elle est un puissant vasodilatateur.
Ceci fonctionne lorsque l’endothélium est normal ; si ce dernier est lésé ou athéromateux, il produit de l’endothéline au lieu de NO• lorsque le stress de paroi augmente [15]. La perte de production de NO• est un des premiers indices de dysfonctionnement dans l’artériosclérose ; les statines ont la capacité d’augmenter cette production [10].
| Contrôle de la pression artérielle |
|
La PA normale maximale est 130/80 mmHg; la PA différentielle normale maximale est 60 mmHg. Hypertension artérielle: PA > 140/80, PAdiff > 80 mmHg.
Le sinus carotidien et les barorécepteurs aortiques sont stimulés par la distension de la paroi artérielle et par la pulsatilité; ils déclenchent une stimulation vagale (hypotension et bradycardie).
Le glomus carotidien est stimulé par l'hypoxie; il déclenche une stimulation sympathique.
La distension des parois auriculaires provoque la sécrétion d'ANF (diurétique et bradycardisant).
La distension des parois ventriculaires provoque la sécrétion de BNP (diurétique et vasodilatateur).
La baisse brusque de la précharge induit le réflexe de Bezold-Jarish (bradycardie, vasodilatation artérielle); en cas d'hypovolémie, il associe une bradycardie à l'hypotension.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- BIAGGIONI I, SHIBAO CA, DIEDRICH A, et al. Blood pressure management in afferent baroreflex failure. J Am Coll Cardiol 2019; 74:2939-47
- CRYSTAL GJ, SALEM MR. The Bainbridge and the “reverse” Bainbridge reflexes: History, physiology and clinical relevance. Anesth Analg 2012; 114:520-32
- EBERT TJ. Autonomic balance and cardiac function. Curr Opin Anesthesiol 1992; 5:3-10
- FARAG E, MAHESHWARI K, MORGAN J, et al. An update of the role of rennin angiotensin in cardiovascular homeostasis. Anesth Analg 2015; 120:275-92
- GALLEY HF, WEBSTER NR. Physiology of the endothelium. Br J Anaesth 2004; 93:105-13
- JASON AC, CARTER C. Clinical relevance of the Bezold-Jarisch reflex. Anesthesiology 2003; 98:1250-60
- KEMP M, DONOVAN J, HIGHAM H, HOOPER J. Biochemical markers of myocardial injury. Br J Anaesth 2004; 93:63-73
- LEVICK JR. An introduction to cardiovascular physiology. 2nd edition. Oxford, Butterworth-Heinemann, 1995, 255-75
- MARTI CN, GHEORGHIADE M, KALOGEROPOULOS AP, et al. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol 2012; 60:1455-69
- MASUMOTO A, HIROOKA Y, HIRONAGA K, et al. Effect of pravastatin on endothelial function in patients with coronary artery disease (cholesterol-independent effect of pravastatin). Am J Cardiol 2001; 88:1291-4
- MUSTAFA HI, FESSEL JP, BARWISE J, et al. Dysautonomia. Anesthesiology 2012; 116:205-15
- O’ROURKE MF, SAFAR ME. Relationship between aortic stiffening and microvascular disease in brain and kidney. Cause and logic of therapy. Hypertension 2005; 46:200-4
- PATTERSON DJ. Nitric oxide and the autonomic regulation of cardiac excitability. Exp Physiol 2001; 86:1-9
- ROBERTSON D, HOLLISTER AS, FORMAN MB, et al. Reflexes unique to myocardial ischaemia and infarction. J Am Coll Cardiol 1985; 5:99-104
- SCHIFFRIN EL. Endothelin: potential role in hypertension and vascular hypetrophy. Hypertension 1995; 25:1135-43
- SHAPIRO BP, CHEN HH, BURNETT JC, et al. Use of brain natriuretic peptide to aid in the diagnosis of heart failure. Mayo Clin Proc 2003; 78:481-6
- SOWERS JR. Hypertension, angiotensin II and oxidative stress. N Engl J Med 2002; 346:1999-2001
- VASAN RS, LARSON MG, LEIP EP, et al. Impact of high-normal blood pressure on the risk of cardiovascular disease. N Engl J Med 2001; 345:1291-7
- WHELTON PK, CAREY RM, ARONOW WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/AphA/ASH/ASPC/NMA/ PVNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive summary. J Am Coll Cardiol 2018; 71:2199-269
- YASTREBOV K. Intraoperative management: carotid endarterectomies. Anesthesiol Clin N Am 2004; 22:265-87
- ZUCKER IH, CORNISCH KG. The Bezold-Jarisch reflex in the conscious dog. Circ Res 1981; 49:940-8