Alors que l'hypoxie est un apport insuffisant d'oxygène sans diminution de la perfusion tissulaire, l'ischémie se caractérise par une baisse de la perfusion entraînant un manque d'oxygène et de substrats métaboliques, et une élimination incomplète des métabolites. Elle représente une souffrance tissulaire due à un déséquilibre entre l’apport (DO2) et la demande (VO2) en oxygène (Figure 9.3).
- L’insuffisance dans l’apport d’O2 (supply ischaemia) peut être due à un vasospasme, à une sténose serrée, à une obstruction (thrombose sur une plaque), à une hypotension, à une anémie ou une hypoxie sévères ; elle est le plus souvent responsable du syndrome coronarien aigu. Le myocarde survit à une ischémie sévère par une combinaison d'inhibition de la contraction (hibernation) et de glycolyse anaérobique.
- L’excès dans la demande en O2 (demand ischaemia) est lié à la tachycardie, à l’augmentation de la tension de paroi et à la stimulation sympathique de l’effort, non accompagnées d’une augmentation correspondante du flux coronarien ; cette situation est caractéristique des épisodes d’angor stable chronique.
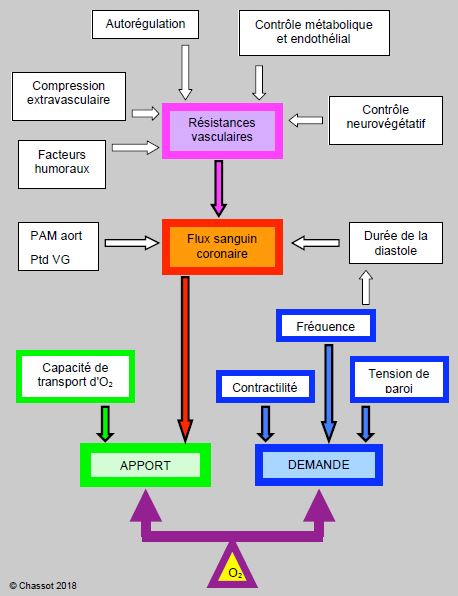
Figure 9.3 : Représentation schématique des facteurs affectant l'équilibre entre l'apport et la demande myocar-dique en oxygène (rapport DO2/VO2). PAM aort: pression artérielle moyenne de l'aorte ascendante. Ptd: pression télédiastolique du VG.

La plaque coronarienne stable est un dépôt athéromateux fixe, fibrosé ou calcifié, occasionnant une sténose serrée (> 75%) et un angor chronique ; cette lésion est bien visible à la coronarographie et entraîne une symptomatologie décelable aux tests d’effort. La plaque instable est un dépôt lipidique très inflammatoire recouvert d’une capsule mince et friable, très susceptible de rupture ; comme elle représente une sténose peu serrée (≤ 50%), son risque est essentiellement lié à son activité inflammatoire, à son degré d’instabilité et à l’agrégabilité plaquettaire. Alors qu’elle est la cause de trois quarts des syndromes coronariens aigus en clinique, la rupture de plaque instable n’est à l’origine que de 45% des infarctus postopératoires ; dans le contexte chirurgical, l’ischémie par déséquilibre DO2/VO2 est plus fréquente (55% des cas) [17].
Des épisodes récurrents d'ischémie peuvent altérer structurellement le myocarde: hypertrophie, fibrose interstitielle, sclérose. La production endothéliale locale de NO• est très affaiblie en cas de sténose coronarienne, alors que la sécrétion d'endothéline et d'angiotensine II est élevée; ces deux substances ont des effets vasoconstricteurs mais aussi potentialisateurs de la fibrose interstitielle. L’angiotensine II est un des agonistes principaux parmi les nombreux déterminants moléculaires de l’HVG, mais les déclencheurs de croissance qui favorisent l’hypertrophie des cellules musculaires sont aussi des facteurs qui activent les fibroblastes et, à partir d’un certain seuil, engendrent l’apoptose [12].
L'angor est causé par la libération d'adénosine. L'angor stable est caractéristique d'une diminution chronique du flux sanguin local par une sténose coronarienne, entraînant une souffrance ischémique distale lorsque la demande en O2 augmente. Dans l'angor instable, une érosion ou une fissure dans une plaque instable mettent le flux sanguin en contact avec les lipides et les macrophages de la plaque. Ceci conduit à une vasoconstriction locale et à l'adhésion des thrombocytes, entraînant la formation d'un thrombus qui peut devenir occlusif ou être spontanément lysé en 10 à 20 minutes [9].
Syndrome coronarien aigu (SCA)
Dans les premières minutes qui suivent une occlusion coronarienne survient une série de modifications qui se déroulent dans l’ordre chronologique suivant (Figure 9.4 et Tableau 9.1) [7,16] :
- Baisse de la compliance ventriculaire (dysfonction diastolique par défaut de relaxation active protodiastolique);
- Altération de la cinétique segmentaire (ACS) à l’échocardiographie;
- Dysfonction systolique et baisse de la tension de paroi en systole;
- Modification du segment ST à l’ECG;
- Baisse de la FE mesurée (si la zone ischémiée est > 15% de la masse ventriculaire);
- Une absence totale de flux sanguin durant > 20 minutes entraîne progressivement la nécrose; la lésion d'infarcissement est irréversible après 2-4 heures.
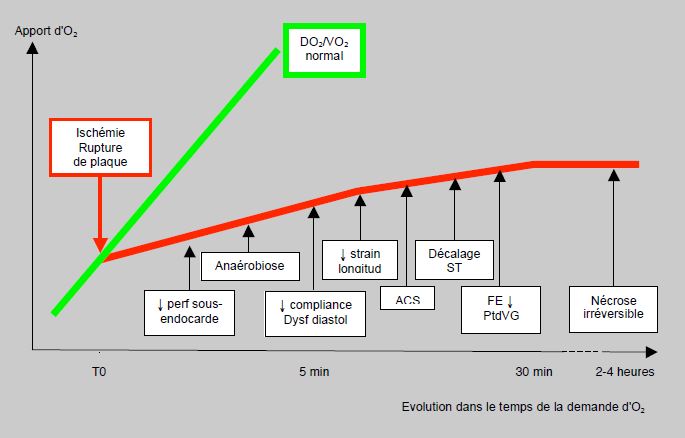
Figure 9.4 : Chronologie des évènements après une interruption du flux coronarien par une rupture de plaque. La dysfonction diastolique survient avant la dysfonction systolique. Les anomalies de la cinétique segmentaire (ACS) surviennent après quelques minutes et précèdent le décalage du segment ST de 30 secondes à 2 minutes. L'angor se manifeste moins d'une minute après l'occlusion coronarienne. Les lésions de nécrose deviennent irréversibles à partir de 2-4 heures. Strain longitud: déformation longitudinale due à la contraction des faisceaux myocardiques sous-endocardiques. PtdVG: pression télé-diastolique du VG. FE: fraction d'éjection [10,16].
La dysfonction segmentaire échocardiographique apparaît 1-2 minutes avant les signes électriques. Il faut une diminution de ≥ 60% du flux pour induire une hypokinésie et de ≥ 80% pour provoquer une akinésie ; ces altérations de la cinétique segmentaire sont potentiellement réversibles par revascularisation. Une hypokinésie fixe apparaît à l’échocardiographie dans le territoire concerné lorsque > 20% de l’épaisseur de paroi est infarcie ; il faut un infarcissement de > 40% de l’épaisseur pour provoquer une akinésie (Vidéos). La baisse de la tension de paroi systolique atteint son nadir à la vingtième minute. La zone ischémiée présente un mouvement paradoxal sous forme d‘un raccourcissement et d’un épaississement post-systoliques qui ne contribuent pas à l’éjection du sang puisqu’ils ont lieu après la fermeture de la valve aortique (voir Figure 9.14) ; par contre, ils perturbent la relaxation protodiastolique. Les lésions de nécrose deviennent irréversibles à partir de 2-4 heures, d'où la nécessité de revasculariser dans les 2 premières heures après le début de la crise d'angor. La lésion myocardique est définie par une élévation des troponines > 99ème percentile de la limite supérieure de référence; la lésion est considérée comme aiguë lorsque les troponines s'élèvent ou s'abaissent [23]. Les effets hémodynamiques dépendent de la masse ventriculaire touchée et de sa localisation : la perte de 25% du myocarde entraîne une insuffisance ventriculaire et celle de > 35% conduit au choc cardiogène [16]. Un infarctus antérieur ou latéral a davantage de retentissement hémodynamique qu’une lésion septale ou postérieure, parce que le degré de raccourcissement radiaire en systole est plus élevé dans les parois antérieure et latérale que dans la paroi postérieure et dans le septum (Figure 9.5).
Vidéo: akinésie antéro-apicale avec dilatation de l'apex sur ischémie du territoire de l'IVA en vue mi-oesophagienne 4-cavités 0°.
Vidéo: akinésie antéro-apicale avec dilatation de l'apex sur ischémie du territoire de l'IVA en vue mi-oesophagienne 2-cavités 90°.
Vidéo: akinésie antéro-apicale avec dilatation de l'apex sur ischémie du territoire de l'IVA en vue mi-oesophagienne 4-cavités 0°.
Vidéo: akinésie antéro-apicale avec dilatation de l'apex sur ischémie du territoire de l'IVA en vue mi-oesophagienne 2-cavités 90°.
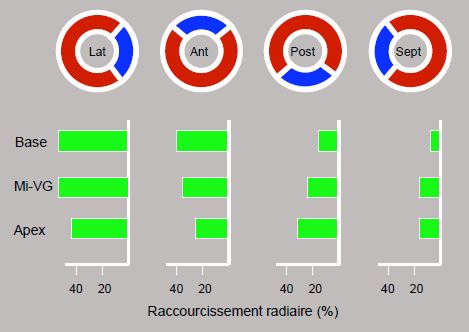
Figure 9.5 : Raccourcissement radiaire selon les 4 quadrants du VG : latéral, antérieur, postérieur et septal. Le degré de contraction est le plus important dans le quadrant latéral, un peu moins dans le quadrant antérieur, et nettement plus faible dans les quadrants postérieur et septal. Le degré de raccourcissement de la paroi postéro-basale est plus faible que le reste du VG, raison pour laquelle elle paraît souvent hypokinétique à l’échocardio-graphie. Ainsi, la contribution de chaque quadrant au volume systolique éjecté est différente; la localisation d'une akinésie determine donc son impact hémodynamique [18,19].
D’un point de vue physiopathologique, les mécanismes en jeu dans les syndromes coronariens aigus peuvent être répartis en cinq catégories (voir Figure 9.2) [7,15].
- Athéromatose obstructive avec réaction inflammatoire systémique ; cette dernière est accompagnée d’une flambée immunitaire et se caractérise par de multiples ruptures de plaques, une activation des neutrophiles, des macrophages et des lymphocytes T, une CRP élevée et une libération locale de cytokines. Un thrombus obstrue la lumière.
- Athéromatose obstructive sans réaction inflammatoire ; des stresseurs physiques (stress mécanique de la paroi artérielle) ou émotionnels déclenchent une réaction sympathique majeure avec hypertension, vasoconstriction locale, activation plaquettaire et augmentation de la mVO2. Les modifications physico-chimiques des plaques (température, pH, fumée, toxines) peuvent conduire à une cristallisation massive de leur cholestérol et à leur éclatement.
- Erosion endothéliale avec perte de cellules endothéliales et infiltration de neutrophiles mais sans implication inflammatoire. Les points ainsi dénudés d'endothélium sont des attracteurs pour les plaquettes et les polymorphonucléaires; il se forme un "thrombus blanc" riche en thrombocytes qui active la thrombine et le dépôt local de fibrine.
- Vasospasme avec athéromatose non-obstructive ; une vascoconstriction intense est présente dans les vaisseaux épicardiques et dans la microcirculation, comme dans le syndrome de Takotsubo. La fonction endothéliale est altérée et induit un vasospasme alors qu’elle devrait produire du NO en réponse à la stimulation sympathique.
- Dissection spontanée d'une coronaire.
La rupture de plaque instable et l'adhésion des plaquettes sur la zone cruentée provoquent la formation d'un thrombus plus ou moins occlusif qui est classiquement considéré comme le substrat physiopathologique commun des syndromes coronariens aigus puisque qu’on le retrouve dans > 75% des SCA, principalement sur des troncs proximaux ou des bifurcations [3,7]. Mais de nombreuses investigations ont montré la présence de thrombus sur des plaques fissurées sans pour autant que les patients ne présentent de symptômes ischémiques. Il s’ensuit que l’origine du SCA est certainement multifactorielle, résultant de la combinaison malheureuse d’un état inflammatoire, d’une hypercoagulabilité, d’un pic dans l’effet de toxines (lipides, fumée, polluants) ou dans la réponse au stress (catécholamines, hypertension), synchronisés avec le moment d’une rupture ou d’une fissure dans une plaque instable située à un endroit critique du réseau coronarien [4].
La position du segment ST à l'ECG occupe une place centrale dans la définition clinique du syndrome coronarien aigu (Figure 9.6). Il introduit une dichotomie fondamentale entre le sus-décalage et le sous-décalage ST [1,2,3,4].
- Un quart des patients souffrant de syndrome coronarien aigu présente une surélévation du segment ST ; la majorité d'entre eux développe un infarctus avec onde Q (ST-elevation myocardial infarction ou STEMI) ; un thrombus coronarien occlusif est présent dans > 80% des cas.
- Les trois autres quarts présentent un sous-décalage ST et souffrent en majorité d'angor instable ; certains développeront un infarctus (élévation des troponines), en général de type non-Q (non-STEMI) ; un thrombus coronarien n’est présent que dans 35-60% des cas et n'est pas occlusif.
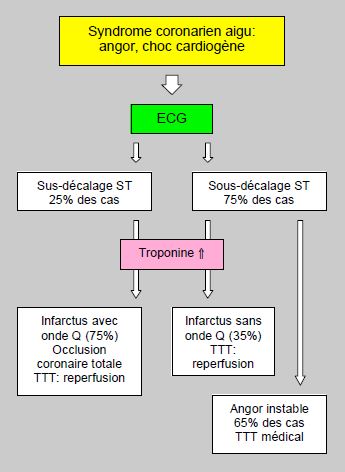
Figure 9.6 : La position du segment ST à l'ECG occupe une place centrale dans la définition du syndrome coronarien aigu. L'élévation des troponines et le décalage ST certifient le diagnostic d'infarctus. La majorité des patients avec un sus-décalage ST développe un infarctus avec onde Q (STEMI). Les patients qui présentent un sous-décalage ST souffrent en majorité d'angor instable; certains développent un infarctus (élévation des troponines), en général de type non-Q (N-STEMI). La mortalité (environ 5%) est pratiquement la même pour des deux types d'infarctus [3].
La mortalité (en moyenne 5%) est un peu plus élevée pour les infarctus de type STEMI. C'est l'élévation du taux d'enzymes cellulaires, essentiellement les troponines, qui certifie le diagnostic d'infarctus en cas de décalage du segment ST. Les patients qui présentent une surélévation persistante du segment ST sont candidats à une technique de reperfusion en urgence (thrombolyse ou angioplastie percutanée), alors que ceux qui ont une sous-dénivélation ST peuvent bénéficier d'un traitement médical anti-ischémique, suivi plus ou moins rapidement d'une revascularisation (voir Traitement, Ischémie aiguë). Les antiplaquettaires font partie du traitement aigu des deux catégories de malades [1,3,5,14,20,21,22].
| Ischémie myocardique |
|
L'ischémie est une souffrance tissulaire due à un déséquilibre entre l’apport (DO2) et la demande (VO2) en oxygène:
- La baisse de DO2 peut être due à: vasospasme, sténose serrée, obstruction par thrombose, hypotension, anémie, hypoxie
- L'excès de VO2 est lié à: tachycardie, augmentation de tension de paroi et de contractilité, stress, douleur
L'ischémie entraîne dans les 20 minutes les évènements suivants, par ordre chronologique:
- Angor (dû à la libération d'adénosine)
- Dysfonction diastolique
- Altération de la cinétique segmentaire
- Dysfonction systolique
- Altérations ST (ECG)
Une interruption totale du flux de > 20 minutes provoque la nécrose cellulaire (en l’absence de collatérales); la lésion tissulaire devient irréversible après 2-4 heures. Les effets hémodynamiques dépendent de la masse ventriculaire lésée : 25% entraînent une insuffisance ventriculaire et > 35% un choc cardiogène.
Le syndrome coronarien aigu est défini par la position du segment ST: sus-décalage (infarctus STEMI) ou sous-décalage (infarctus non-STEMI). L’infarctus est défini par l’élévation des troponines.
- Angor + sus-décalage ST + troponines élevées: infarctus STEMI
- Angor + sous-décalage ST + troponines élevées: infarctus N-STEMI
- Amgor + sous-décalage ST sans troponines: angor instable
|
L'infarctus
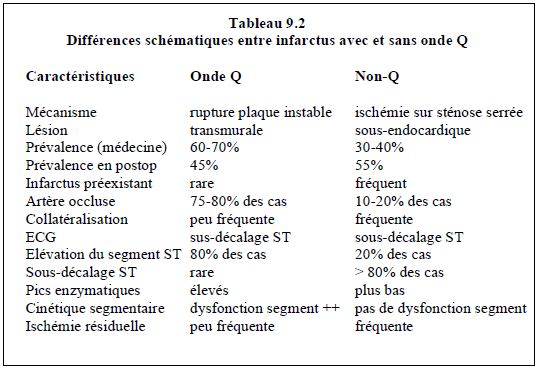
En l'absence de collatérales, une interruption de flux sanguin pendant une vingtaine de minutes entraîne la nécrose cellulaire. Le phénomène est accéléré si la mVO2 est élevée ou la pression artérielle basse. En revanche, le délai est étendu à 2 - 6 heures si le réseau collatéral est bien développé. La nécrose débute habituellement dans la zone sous-endocardique et s’étend progressivement vers l'épicarde pour devenir transmurale en 4 à 6 heures. Le délai de 2 heures représente les golden hours pendant lesquelles on peut interrompre le processus en reperméabilisant le vaisseau concerné (voir Traitement de l’ischémie aiguë). On peut distinguer deux types d'infarctus selon qu’on est en présence d’une plaque stable ou d’une plaque instable (Tableau 9.2). Il existe 5 différents types d'infarctus classés selon le contexte clinique [2,11,23].
- Type 1: infarctus sur thrombose par rupture ou érosion d'une plaque d'athérome (le plus souvent STEMI). La coronarographie met en évidence l'obstruction responsable de la lésion.
- Type 2: infarctus par déséquilibre DO2/VO2 (le plus souvent N-STEMI); ce déséquilibre survient dans le cadre d'une baisse de l'apport d'O2 (coronaropathie stable, spasme, dissection, embolie, anémie, hypoxémie, hypotension) ou d'une augmentation de la consommation d'O2 (tachycardie, hypertension, arythmie, effort, stress, chirurgie) [8]. La coronarographie ne met en évidence aucune obstruction athéromateuse qui pourrait être à l'origine de la lésion.
- Type 3: mort subite cliniquement de nature ischémique, sans confirmation ECG ni biomarqueurs.
- Type 4: infarctus au cours de PCI (4a), sur thrombose de stent (4b) ou sur resténose (4c); les troponines s'élèvent à ≥ 5 fois la valeur normale supérieure.
- Type 5: infarctus lié à des pontages aorto-coronariens; les troponines s'élèvent à ≥ 10 fois la valeur normale supérieure dans les 48 heures postopératoires.
La mortalité de l'infarctus de type 2 (10-15%) est plus élevée que celle du type 1 (5-10%), essentiellement par excès de mortalité non-cardiaque liée aux comorbidités, comme le prouve le taux identique d'évènements cardiaque à 5 ans (30 versus 33%) [6,8]. Actuellement, la définition universelle de l’infarctus repose sur les points suivants (pour plus de détails, voir Syndrome clinique) [23].
- Elévation et/ou diminution des marqueurs biochimiques (préférentiellement troponine), dont au moins une valeur est au-delà du 99ème percentile de la limite de référence supérieure (URL: upper reference limit).
- Association à au moins un des éléments suivants:
- Symptomatologie d’angor : douleur rétrosternale constrictive durant 20-30 minutes ; douleur épigastrique de même type ; irradiation dans la mâchoire, l’épaule et le bras gauche.
- Modifications ECG : sus-décalage > 1 mm (0.1 mV) ou sous-décalage > 1-2 mm du segment ST ; apparition d’un bloc de branche gauche.
- Développement d’ondes Q pathologiques.
- Nouvelles anomalies de la contraction segmentaire.
- Présence de thrombus intracoronarien (infarctus type 1) ou de thrombose de stent (type 4b) à la PCI ou à l'autopsie.
- Evidence de déséquilibre DO2/VO2 sans lien avec une thrombose (infarctus type 2)
- Décès cardiaque subit accompagné d’une symptomatologie typique mais sans ECG ni valeur connue de troponine.
Le taux de troponine est mesuré dès que possible et répété 3-6 heures plus tard. Il peut rester élevé jusqu'à 2 semaines après l'événement. La valeur-seuil de troponine considérée comme diagnostique pour un infarctus est déplacée à 5 fois le 99ème percentile de la limite de référence supérieure après PCI et à 10 fois après pontages chirurgicaux [23]. L'infarctus est fréquemment silencieux chez les personnes âgées et les diabétiques ainsi que dans le postopératoire ou en réanimation. Les troponines sont spécifiques au cœur, mais non à la maladie; un taux élevé est le marqueur d'une lésion ou d'une nécrose myocardique, mais n'est pas à lui seul synonyme d'infarctus. Il faut ici distinguer la lésion myocardique de l'infarctus. La première est définie par une simple élévation ou fluctuation des troponines au-delà du 99ème percentile de la limite de référence supérieure. Le deuxième fait référence à une élévation majeure des troponines accompagnée d'une évidence clinique d'ischémie myocardique active et de nécrose [11,23]. Même sans infarctus, toute élévation des troponines est associée à une péjoration du pronostic vital.
Lors d'un infarctus, la dysfonction systolique des segments ischémiés est caractérisée par une hypokinésie (baisse de contractilité), une akinésie (absence de contraction) ou une dyskinésie (expansion vers l’extérieur en systole). Elle est partiellement compensée par une hyperkinésie des segments non touchés. D'autre part, la masse myocardique initialement hypo- ou akinétique au moment de l'ischémie aiguë est plus importante que la masse qui va ultérieurement être infarcie, car toute la zone bordante (pénombre) est immobile, alors qu'elle va potentiellement récupérer par la suite. La fraction d'éjection mesurée à un moment proche de l'évènement aigu peut être ainsi inférieure à ce qu'elle sera après récupération. Le degré d'augmentation du volume télésystolique et la présence d'une dysfonction diastolique sont des bons prédicteurs de la mortalité post-infarctus. La gravité d’un infarctus dépend de plusieurs éléments.
- La localisation ; un infarctus antérieur ou latéral a de fortes conséquences hémodynamiques parce que la contraction des parois antérieure et latérale contribue davantage au volume systolique que celle de la paroi postérieure et du septum.
- Le type de lésion ; un infarctus transmural modifie la contractilité d’un ou de plusieurs segments (chaque segment représente environ 5% de la masse du VG), alors qu’un infarctus sous-endocardique peut ne pas altérer significativement la fonction systolique du ventricule. Il faut une lésion impliquant plus de 20% de l'épaisseur de la paroi myocardique pour voir apparaître une altération de la cinétique segmentaire.
- La dimension de la lésion ; plus il est étendu, plus un infarctus pénalise le débit cardiaque. Il faut que plus de 15% de la masse du VG ait une contraction anormale pour que la fraction d'éjection globale soit modifiée. La taille d'un infarctus dépend de plusieurs éléments: la dimension de la zone à risque, le degré de collatéralisation, la durée d'interruption du flux, les conditions hémodynamiques (pression artérielle, fréquence cardiaque) et la température [13].
- Le risque d’arythmies ; la présence de tachy-arythmies auriculaires ou ventriculaires (TV, fibrillation) augmente la mortalité à court terme et aggrave le pronostic à long terme.
- Le degré de collatéralisation ; les sténoses serrées (> 90%) entraînent une ischémie chronique (angor d’effort) qui génère un recrutement de vaisseaux collatéraux, alors que la thrombose aiguë d’une plaque instable (sténose < 60%) est une occlusion brutale de vaisseau non collatéralisé.
- Le risque de complications ; la rupture pariétale et la CIV sont caractéristiques de lésions touchant une artère terminale non collatéralisée.
- L’insuffisance mitrale ; la survenue d’une IM majeure sur ischémie pariétale quadruple le risque d’insuffisance congestive et double la mortalité (voir Chapitre 11, IM secondaire ischémique).
Laissé à son évolution spontanée, un infarctus avec surélévation du segment ST a une mortalité de 30%, dans la moitié des cas pendant les premières heures par fibrillation ventriculaire ; cette mortalité tombe à 5% lorsque le malade est pris en charge agressivement [3]. Lors de la stabilisation et de la cicatrisation, le ventricule change de forme, de taille et d'épaisseur: c'est le remodelage. L'amincissement de la zone infarcie (< 0.6 cm d’épaisseur) entraîne une dyskinésie importante, qui réduit d'autant le volume éjecté. La dilatation du VG est une réponse physiologique à cette perte dans le but de maintenir la performance systolique, mais elle désavantage le ventricule à long terme car sa tension de paroi s’élève, et elle augmente le risque d'arythmies. Le degré d'augmentation du volume télésystolique est un excellent prédicteur de la mortalité post-infarctus. Les modifications géométriques des piliers et de leur position peuvent donner naissance à une insuffisance mitrale (voir ETO des complications ischémiques, Figures 9.20 et 9.21).
| Infarctus myocardique |
|
L’infarctus est une nécrose tissulaire qui survient dès 20 minutes après une occlusion coronarienne totale. En présence de collatérales, ce délai est repoussé à 4-6 heures. Le phénomène est accéléré si la mVO2 est élevée ou la PA systémique basse.
Les altérations de la cinétique segmentaire sont fonction de l’épaisseur de paroi touchée : ≥ 20% provoque une hypokinésie et ≥ 40% une akinésie. La FE baisse si ≥ 15% de la masse du VG est infarcie. En phase aiguë, la zone bordante (pénombre) élargit la taille de l’hypo/akinésie mais est potentiellement récupérable.
On distingue 2 types d'infarctus selon la présence ou non d'une onde Q à l'ECG et selon l'étiologie relevant d’une plaque stable (déséquilibre DO2/mVO2) ou d’une plaque instable (thrombose).
- En clinique cardiologique: 2/3 des infarctus sont dus à une rupture de plaque instable
(sus-décalage ST > 1 mm, infarctus STEMI, présence d’onde Q) ;
- En postopératoire: 50-60% sont dus à un déséquilibre DO2/mVO2 (sous-décalage ST > 2 mm, infarctus non-STEMI).
|
© BETTEX D, CHASSOT PG, RANCATI V, Janvier 2008, dernière mise à jour, Novembre 2019
Références
- ANDERSON JL, ADAMS CD, ANTMAN EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction. Circulation 2007;116:e148-e304
- ANDERSSON JL, MORROW DA. Acute myocardial infarction. N Engl J Med 2017; 376:2053-64
- ANTMAN EM, BRAUNWALD E. ST-elevation myocardial infarction: Pathology, pathophysiology, and clinical features. In: ZIPES DP, et al, eds. Braunwald's heart disease. A textbook of cardiovascular medicine, 7th edition. Philadelphia, Elsevier-Saunders, 2005, 1141-65
- ARBAB-ZADEH A, NAKANO M, VIRMANI R, FOSTER V. Acute coronary events. Circulation 2012; 125:1147-56
- ARNST HR, BOSSAERT LL, DANCHIN N, NIKOLAOU NI. European Resuscitation Council Guidelines for resuscitation 2010. Section 5. Initial management of acute coronary syndrome. Resuscitation 2010; 81:1333-63
- CHAPMAN AR, SHAH ASV, LEE KK, et al. Long-term outcomes in patients with type 2 myocardial infarction and myocardial injury. Circulation 2018; 137:1236-45
- CREA F, LIBBY P. Acute coronary syndromes. The way forward from mechanisms to precision treatment. Circulation 2017; 136:1155-66
- DEFILIPPIS AP, CHAPMAN A, MILLS NL, et al. Assessment and treatment of patients with type 2 myocardial infarction and acute nonischmeic myocardial injury. Circulation 2019; 140:1661-78
- FUSTER V, FAYAD ZA, BADIMON JJ. Acute coronary syndromes: biology. Lancet 1999; 353(suppl III):5-9
- GARG P, UNDERWOOD R, SENIOR R, et al. Noninvasive cardiac imaging in suspected acute coronary syndrome. Nat Rev Cardiol 2016; 13:266-75
- GOEDDEL LA, HOPKINS AN, FERNANDO RJ, et al. Analysis of the 4th Universal Definition of Myocardial Infarction – Key concepts and perioeprative implications. J Cardiothorac Vasc Anesth 2019; 33:3486-95
- HEIN S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart. Circulation 2003; 107:984-91
- IBÁÑEZ B, HEUSCH G, OVIZE M, et al. Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 2015; 65:1454-71
- IBANEZ B, JAMES S, AGEWALL S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 2018; 39:119-77
- KANWAR SS, STONE GW, SINGH M, et al. Acute coronary syndromes without coronary plaque rupture. Nat Rev Cardiol 2016; 13:257-65
- KERN MJ. Coronary blood flow and myocardial ischemia. In: ZIPES DP, et al. Braunwald’s Heart disease. A textbook of cardiovascular medicine. 7th edition Philadelphia: Elsevier Saunders, 2005, 1103-28
- LANDESBERG G. The pathophysiology of perioperative myocardial infarction: Facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90-100
- LORENZ CH, PASTOREK JS, BUNDY JM. Delineation of normal human left ventricular twist throughout systole by tagged cine magnetic resonance imaging. J Cardiovasc Magn Reson 2000; 2:97-108
- MAIER SE, FISCHER SE, McKINNON GC, et al. Evaluation of left ventricular segmental wall motion in hypertrophic cardiomyopathy with myocardial tagging. Circulation 1992; 86:1919-28
- O’CONNOR RE, BRADY W, BROOKS SC, et al. 2010 AHA Guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Part 10: acute coronary syndromes. Circulation 2010; 122(suppl 3):S787-S817
- O’GARA PT, KUSHNER FG, ASCHEIM DD, et al. 2013 ACCF/AHA Guideline for the management of ST-elevation myocardial infarction. Circulaion 2013; 127: e362-e425
- STEG G, JAMES SK, ATAR D, et al. ESC Guidelines for the management of acute myocardial infarction in pratients presenting with ST-segment elevation. Eur Heart J 2012; 33:2569-619
- THYGESEN K, ALPERT JS, JAFFE AS, et al. Fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol 2018; 72:2231-64