L’ischémie
Lorsque l’apport d’O2 est interrompu, la chaîne respiratoire mitochondriale cesse de fonctionner après une minute, la glycolyse passe en mode anaérobique et l’utilisation des acides gras est stoppée. Le pH intracellulaire commence à chuter et le taux de lactate à monter. En quinze minutes d'ischémie normothermique, l’ATP myocardique baisse de 65%. Vingt minutes suffisent à entraîner des foyers de nécrose myocardique irréversible; ceci survient lorsque le taux d’ATP est < 0.2 nmol/mg [2]. Bien qu’elle puisse fonctionner pendant une quarantaine de minutes, la glycolyse anaérobique ne peut que maintenir le fonctionnement des pompes ioniques, mais non assurer la contraction myocardique; le myocarde ne reste viable que s’il est arrêté. S'il est reperfusé à ce stade, il peut récupérer [7].
Lorsque les pompes dysfonctionnent à leur tour, le Na+ rentre massivement dans la cellule à cause de la chute d’activité des Na+/K+-ATPases et de l’échangeur Na+/H+ qui extrait les valences acides ; cette surcharge sodique provoque un œdème cellulaire. De son côté, le Ca2+ n’est plus repompé par le reticulum sarcoplasmique (RS) par manque d’ATP et s’accumule dans le cytoplasme, où il active les protéases, les lipases et les nucléases [2]. Les canaux MPTP de la paroi mitochondriale (mitochondrial permeability transition pore), qui sont normalement fermés, s’ouvrent lors d’une souffrance ischémique extrême. Cette ouverture signe l’arrêt de mort de la mitochondrie, car elle perd son imperméabilité : elle gonfle, son potentiel de membrane s’effondre, ses cristae se rompent, et ses composants propres comme le cytochrome c et les radicaux libres sont relargués dans la cellule, où ils activent les caspases (enzymes digérant les composants cellulaires) (Figure 24.1) [16]. Le cycle de Krebs s'arrête et la cellule meurt.
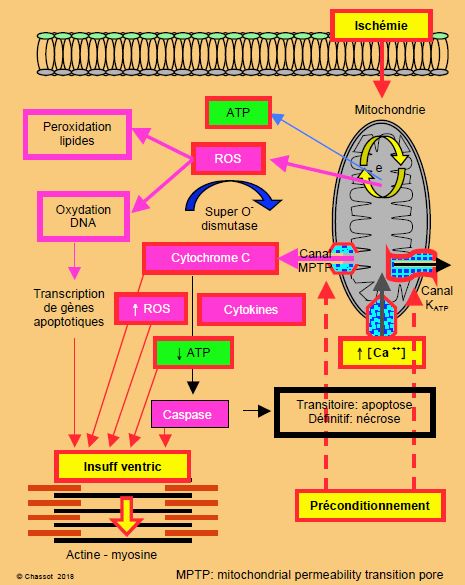
Figure 24.1 : Schématisation des lésions de l'ischémie. La mitochondrie laisse fuir (par le canal MPTP) des agents oxydants comme le cytochrome C, les ROS (Reactive Oxygen Species) ou superoxydes et des cytokines, qui vont freiner la synthèse d’ATP et activer les caspases qui digèrent la cellule (voir description détaillée au Chapitre 5, Apoptose). e• : chaîne d’oxydo-réduction de la mitochondrie. MTPT : mitochondrial permeability transition pore. Le préconditionnement prévient partiellement l’apoptose en agissant sur deux points au niveau des mitochondries : 1) ouverture des canaux potassiques dépendants de l’ATP (KATP), ce qui diminue la charge en calcium ionisé (Ca2+) dans le cytosol mitochondrial, et 2) fermeture des canaux de perméabilité MPTP, ce qui diminue le taux de caspases cytoplasmiques [d'après réf 16].
La reperfusion
Au lieu de rétablir la situation, la reperfusion du myocarde ischémié génère des lésions supplémentaires qui s’ajoutent à celles induites par l’interruption circulatoire, et met en route une cascade de phénomènes pouvant aboutir à des dégâts cellulaires irréversibles [2,8,15]. Elle cause un oedème explosif de la cellule et des organelles intracellulaires à cause du gradient osmotique massif attirant l'eau et les électrolytes (Na+, Ca2+) vers l'intérieur de la cellule. L'oedème cellulaire, la lésion des membranes et la libération d'enzymes cytotoxiques sont d'autant plus importants que le perfusat contient davantage d'oxygène et de calcium [13]. Alors que l'hypothermie a un rôle protecteur pendant la phase ischémique, elle n'en a aucun pendant la phase de reperfusion. Toute une série de processus se met en route pendant cette période (Tableau 24.1, Figure 24.2) [7].

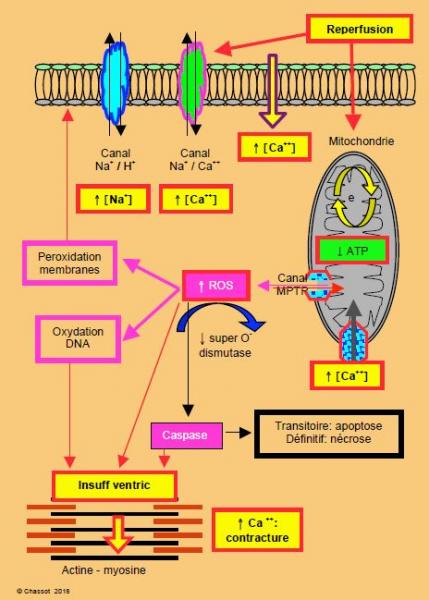
Figure 24.2 : Schématisation des lésions de reperfusion. La réactivation des canaux échangeurs Na+/H+ et Na+/Ca++ augmentent la concentration intracellulaire de Na+ et de Ca++. Les membranes laissent passer le Ca++. L'ouverture du canal MPTP (mitochondrial permeability transition pore) laisse entrer les cations dans la mitochondrie et sortir des agents oxydants comme le cytochrome C et les ROS (Reactive Oxygen Species), qui vont activer les caspases (digestion de la cellule), car les enzymes antioxydants font défaut après une période d'ischémie. e• : chaîne d’oxydo-réduction de la mitochondrie.
- L’apport soudain d’O2 dans un milieu riche en radicaux libres va donner naissance à des produits très toxiques, les ROS (reactive oxygen species); ceux-ci ne sont plus dégradés car l'ischémie a réduit les défenses naturelles contre ces superoxydes (superoxyde dismutase, réductases, etc).
- L'arrivée d'O2 dans une cellule démunie de ses défenses altère les membranes qui deviennent perméable au Ca2+.
- L’activation des canaux calcique lents (canaux L) et de l’échangeur Na+/Ca2+ qui extrait le Na+ fait rentrer une grande quantité de Ca2+ dans la cellule.
- La flambée de ROS ouvre les canaux MPTP (mitochondrial permeability transition pore) de la paroi mitochondriale qui étaient restés fermés pour protéger la mitochondrie de l'acidose ischémique; les cations pénètrent ainsi dans la mitochondrie et découplent la chaîne d’oxydo-phosphorylation; la production d'ATP s'effondre.
- Les mitochondries utilisent l'oxygène pour le repompage du Ca2+ excédentaire et non pour la formation d'ATP. L'excès de Ca2+ y induit la formation de caspases, enzymes digérant les protéines cellulaires, qui fuient dans le cytoplasme par les canaux MTPT.
- La rigidification des myofibrilles (stone heart) due au blocage des ponts actine-myosine est secondaire à l’excès de Ca2+ libre dans le cytoplasme.
- La production soudaine et excessive de NO• lève l’inhibition sur la transmigration leucocytaire et sur la régulation des superoxydes.
Il existe donc un paradoxe de l'oxygène et du calcium: alors que leur manque conduit à la nécrose ischémique, leur apport soudain à la cellule qui en a été privée déclenche une cascade d'évènements pathologiques qui sont plus graves que ne l’étaient les dégâts de l’ischémie. La reperfusion après un épisode ischémique est supposée guérir les lésions myocardiques ; or elle contribue à l’aggravation des dégâts cellulaires secondaires à l’occlusion vasculaire et compte pour moitié dans la taille finale de l’infarctus [7,8,15].
En clinique, on rencontre cinq types de lésions de reperfusion.
- La sidération myocardique (stunning), ou dysfonction contractile aiguë sans lésion cellulaire apparente, qui survient après rétablissement d’un flux coronaire normal.
- La rigidification des myofibrilles par l'excès de Ca2+, qui aboutit au "cœur de pierre" non-contractile (stone heart). Le ventricule est contracté et rigide, prompt à la fibrillation, et incapable de soutenir la circulation malgé des coronaires peméables.
- Le phénomène de no-reflow, ou incapacité de revasculariser une zone ischémique à cause d’une impédance microvasculaire excessive; alors que les vaisseaux épicardiques sont bien perméables, les capillaires sont obstrués par des microthromboses, des dégâts endothéliaux et des cellules oedémateuses. A l'angiographie, le flux distal est lent ou absent alors que les gros vaisseaux sont perméables. Ce phénomène survient dans 10-30% des STEMI revascularisés et peut durer jusqu'à une semaine [12]. La persistance d'une zone de non-reperfusion est associée à la dilatation secondaire du ventricule [10].
- Les arythmies de reperfusion, le plus souvent des tachy-arythmies ventriculaires itératives difficile à juguler. Lorsque le rythme est régulier, l'ECG montre des ondes T inversées, amples et pointues typiques des lésions de reperfusion.
- Les lésions léthales, qui entraînent directement la mort cellulaire.
Des interventions thérapeutiques ciblées sur les mécanismes qui entraînent les dégâts au moment de la reperfusion ont un impact sur la taille des infarctus en expérimentation animale, mais n’ont malheureusement aucune application clinique efficace [7]. C’est notamment le cas des perfusions de magnésium, d'anticalciques, d'antioxydants et d'autres substances actives sur les différents canaux transmembranaires (ranolazine, ciclosporine A), dont les résultats sont inconsistants [1,10]. D’autres possibilités de protection ont été envisagées sans plus de succès dans la pratique clinique, comme les peptides natriurétiques et les statines à haute dose qui agissent par l’activation des protéines-kinases accélérant la recapture de Ca2+ par le réticulum sarcoplasmique. La perfusion de glucose-insuline-potassium (GIK), tombée en désuétude depuis de nombreuses années, a fait l'objet de nouvelles investigations comme stratégie protectrice du métabolisme myocardique.
- Administrée en préhospitalier en cas de syndrome coronarien aigu, la perfusion de GIK diminue le taux de mortalité et d'arrêt cardiaque par rapport au placebo dans une série de 871 patients, mais elle ne modifie pas le taux d'infarctus [4].
- Après RVA ou PAC, la perfusion de GIK atténue la dysfonction post-CEC (OR 0.41) et diminue les complications cardiovasculaires (OR 0.69) par rapport au placebo dans une série randomisée de 222 patients à haut risque [5].
- L'effet d'une perfusion de GIK sur 1004 cas de STEMI est neutre par rapport au groupe contrôle (976 cas) en terme de mortalité, de choc cardiogène, d'arrêt cardiaque ou de réinfarctus [11].
- L'entretien de la normoglycémie (100-120 mg/dL) sous perfusion insulinique continue (5 UI/kg/min) et adaptation de la perfusion de glucose 20% réduit la morbi-mortalité après chirurgie en CEC (OR 0.62) par rapport à la prise en charge standard, mais elle fait courir le risque d'hypoglycémie (14% des cas) et oblige à doser la glycémie toutes les 10-15 minutes [14].
Les résultats sont donc mitigés et pas encore convaincants. En dépit de ces travaux, seuls le préconditionnement, le postconditionnement et peut-être le conditionnement à distance offrent une protection relativement efficace pour l'instant (voir Préconditionnement). Le manque d'impact clinique des nombreuses tentatives de protection myocardique tiennent probablement à la multiplicité, à l'interdépendance et à la redondance des processus en cause. En viser un seul est courir à l'échec. Il faudrait agir de manière additive au niveau moléculaire, au niveau des mécanismes de transmission (signaling pathways), au niveau cellulaire des cardiomyocytes et au niveau endothélial microvasculaire, de manière à ce que la sommation des effets se traduise par une protection pour le tissu cardiaque. D'autre part, le status clinique du patient (diabète, âge, etc) et ses médications interfèrent avec les différents éléments et compliquent l'interprétation des données [3]. En chirurgie cardiaque, la situation est privilégiée, puisqu'il est possible de protéger le myocarde avant la lésion ischémique, comme avec la cardioplégie. Le point le plus important en clinique, néanmoins, serait d'agir au moment où survient l'infarctus, en disposant de techniques qui bloquent l'installation de la nécrose et qui minimisent les lésions de reperfusion au moment de la revascularisation.
Le calcium
L'élévation massive de la concentration de Ca2+ intracelllulaire [Ca2+]i lors de la reperfusion s'accompagne d'une fuite de Na+ et de K+. Normalement, l'équilibre transmembranaire est maintenu par plusieurs systèmes de pompes actives (voir Figure 5.2 et Figure 5.3).
- Echange Na+/Ca2+: à raison de 3 Na+ pour 1 Ca2+, ce système est le principal moyen pour extraire le calcium de la cellule.
- Echange Na+/K+: cette pompe consommant de l'ATP (ATP sensitive) est la principale source du maintien de la faible concentration en Na+ et de la haute concentration en K+ intracellulaires.
- Normalement, une pompe sensible à l'ATP extrait le Ca2+ libre intracellulaire et le repompe dans le réticulum sarcopolasmique (RS), de sorte que la [Ca2+]i libre reste très basse; ce système est inhibé par l'ischémie. Lors de la reperfusion, la haute [Ca2+]i libre active le repompage par le SR et par les mitochondries, qui y dépensent toute leur énergie au détriment de la synthèse de l'ATP. Le Ca2+ repompé s'accumule dans le SR.
- Echange Na+/H+: ce moyen permet de maintenir le pH intracellulaire en expulsant l'excès des ions acides H+; stimulé par l'ischémie et l'excès de valences acides, ce système accumule du Na+ intracellulaire, qu'il faut ensuite échanger avec du Ca2+, ce qui aggrave l'accumulation de Ca2+i libre.
- Canaux potassiques dépendant de l'ATP (canaux KATP): ils maintiennent le va-et-vient de K+ lors de la stimulation électrique et sont inhibés par l'ischémie et le manque d'ATP. Ils laissent alors fuir le K+, et la membrane devient hyperpolarisée, ce qui est la situation de repos du cardiomyocyte; dans cette situation, les canaux sodiques et calciques sont fermés.
Les radicaux libres
Comme lors du déclenchement de la réaction inflammatoire systémique, la reperfusion et le retour de l'oxygène donne naissance à un excès de radicaux libres (ROS: reactive oxygen species). Ces substances dotées d'un nombre impair d'électrons dans leur couche externe sont très réactives; ce sont essentiellement les radicaux superoxydes (O2•-), le monoxyde d’azote (NO•), le peroxyde d’hydrogène (H2O2) et les peroxynitrites (ONOO-). En faibles quantités, ces molécules sont capitales pour les régulations intracellulaires et l’élimination des déchets moléculaires ; elles fonctionnent aussi comme déclencheur de la cascade du préconditionnement. Elles sont normalement détoxifiées au fur et à mesure de leur production par des anti-oxydants naturels comme la superoxyde dismutase ou les catalases. Cependant, ces systèmes d’élimination, déjà inhibés par l'ischémie, sont vite dépassés lorsqu’elles sont produites en masse lors d’ischémie et de reperfusion. Elles attaquent alors les phospholipides des membranes, les protéines et l’ADN. Elles sont impliquées dans la dysfonction contractile post-ischémique (sidération ou stunning), l’apoptose, la mort cellulaire myocardique, les arythmies et la dysfonction endothéliale de l’athéromatose (voir Chapitre 5 Croissance et apoptose) [9].
La reperfusion entraîne encore des altérations profondes de la vasomotricité coronarienne par diminution du NO• produit localement par l'endothélium. Comme le NO• inhibe l'adhésion des plaquettes et des polymorphonucléaires, sa réduction favorise l'agrégation leucocytaire et plaquettaire, phénomène qui s'accompagne d'une vasoconstriction locale intense. Il existe une relation étroite entre la diminution de la production de NO• et l'importance du flux au moment de la reperfusion [5].
| Ischémie et reperfusion myocardiques |
|
Une minute après l’interruption de l’apport d’O2, le taux d’ATP s’effondre et la glycolyse passe en mode anaérobique. Celui-ci ne permet que le travail des pompes ioniques, mais non la contraction musculaire. Les foyers de nécrose ischémique aparaissent après 20 minutes d’arrêt circulatoire local.
La reperfusion est caractérisée par l’apport soudain d’oxygène et de calcium, ce qui déclenche une cascade d'évènements pathologiques plus graves que ne l’étaient les dégâts de l’ischémie: formation massive de peroxydes (radicaux libres, ROS reactive oxygen species), accumulation de Ca2+ dans le cytoplasme et dans les mitochondries, fuite de Na+ et de K+. La gravité des lésions de reperfusion est proportionnelle à celle des lésions ischémiques qui les précèdent et au contenu en O2 et en Ca2+ du sang qui reperfuse le myocarde.
|
© CHASSOT PG, Juin 2008, dernière mise à jour, Décembre 2019
Références
- ANTMAN E, COOPER H, McKINLEY S, et al. Early administration of intravenous magnesium to high-risk patients with acute myocardial infarction in the Magnesium in Coronaries (MAGIC) trial : a randomized controlled trial. Lancet 2002 ; 360 :1189-96
- BENHABBOUCHE S, CROLA DA SILVA C, FERRARA AR. Base des phénomènes d’ischémie reperfusion et de la protection myocardique. Ann Fr Anesth Réan 2011; 30:S2-S16
- DAVIDSON SM, FERDINANDY P, ANDREADOU I, et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury. J Am Coll Cardiol 2019; 73:89-99
- DUNCAN AE, SESSLER DI, SATO H, et al. Hyperinsulinemic normoglycemia during cardiac surgery reduces a composite of 30-day mortality and serious in-hospital complications. Anesthesiology 2018; 128:1125-39
- ELLENBERGER C, SOLOGASHVILI T, KREIENBÜHL L, et al. Myocardial protection by glucose-insulin-potassium in moderate- to high-risk patients undergoing elective on-pump cardiac surgery: a randomized controlled trial. Anesth Analg 2018; 126:133-41
- ENGELMAN DT, WATANABE M, ENGELMAN RM, et al. Constitutive nitric oxide release is impaired after ischemia and reperfusion. J Thorac Cardiovasc Surg 1995; 110:1047-53
- FERRARI R, BALLA C, MALAGU M, et al. Reperfusion damage – A story of success, failure, and hope. Circ Res 2017; 81:131-41
- FRÄSSDORF J, DE HERT S, SCHLACK W. Anaesthesia and myocardial ischaemia/reperfusion injury. Br J Anaesth 2009; 103:89-98
- KEVIN LG, NOVALIJA E, STOWE DF. Reactive oxygen species as mediators of cardiac injury and protection: the relevance to anesthesia practice. Anesth Analg 2005; 101:1275-87
- KLONER RA, BROWN DA, CSETE M, et al. New and revisited approaches to preserving the reperfused myocardium. Nat Rev Cardiol 2017; 14:67993
- MEHTA SR, YUSUF S DIAZ R, et al. Effect of glucose-insulin-potassium infusion on mortality in patients with acute ST-segment elevation myocardial infarction : the CREATE-ECLA randomized controlled trial. JAMA 2005 ; 293 :437-46
- NICCOLI G, BURZOTTA F, GALIUTO L, et al. Myocardial no-reflow in humans. J Am Coll Cardiol 2009; 54:281-92
- RINNE TT. Cardioprotection and cardioplegia. In: THYS DM, et al, eds. Textbook of cardiothoracic anesthesiology. New York, McGraw-Hill Co, 2001, 488-511
- SELKER HP, BESHANSKY JR, SHEEHAN PR, et al. Out-of-hospital administration of intravenous glucose-isulin-potassium in patients with suspected acute coronary syndromes: the IMMEDIATE randomized controlled trial. JAMA 2012; 307:1925-33
- YELLON DM, HAUSENLOY DJ. Myocardial reperfusion injury. N Engl j Med 2007 ; 357 :1121-35
- ZAUGG M, LUCHINETTI E, UECKER M, et al. Anaesthetics and cardiac preconditioning: signaling and cytoprotective mechanisms. Part I. Brit J Anaesth 2003 ; 91:551-65