Lorsque la valvulopathie entraîne une baisse du débit systolique effectif, le coeur dispose de trois mécanismes compensatoires.
- Augmentation de l'activité sympatho-adrénergique (catécholamines alpha et béta, angiotensine II, aldostérone, ADH) : la vasoconstriction artérielle et la tachycardie permettent de maintenir le débit cardiaque. Les valeurs de pression et de fréquence du patient au repos sont probablement le meilleur compromis hémodynamique possible dans sa situation.
- Effet Frank-Starling : augmentation de la précharge et du volume circulant (rétention d'eau et de sel). La dilatation rend le ventricule plus sphérique, ce qui est pathologique mais présente un avantage mécanique momentané, parce que le raccourcissement circonférentiel des fibres pour éjecter le même volume est moindre. Ce mécanisme est surtout utilisé dans les lésions aiguës ou dans les régurgitations. Le retentissement en amont est une augmentation de la pression auriculaire et une stase (périphérique ou pulmonaire).
- Hypertrophie : en amont d'une sténose valvulaire, la chambre cardiaque s'hypertrophie pour assurer la propulsion du sang malgré l'obstacle à l'éjection (hypertrophie concentrique du VG dans la sténose aortique); en aval, la cavité est de taille réduite (petit VG de la sténose mitrale). Une insuffisance valvulaire retentit également sur la dimension des deux chambres d'amont et d'aval : le ventricule doit accommoder un volume de sang augmenté à chaque battement (dilatation) et accroître son volume systolique pour maintenir le débit antérograde (hypertrophie excentrique) ; la chambre de réception de la régurgitation se dilate également (oreillette dilatée en cas d’IM ou d’IT).
L’anatomie fonctionnelle, telle que l’ETO la met en évidence dans une image des 4 cavités, est une excellente illustration des remodelages et des compensations mises en place par l’organisme pour maintenir son équilibre hémodynamique. Elle permet de se rendre compte des contraintes dominantes et de leurs conséquences. Ainsi, une surcharge de pression (sténose aortique) provoque une hypertrophie ventriculaire gauche concentrique qui se traduit par un VG épais dont la cavité est petite, alors qu’une surcharge de volume (insuffisance mitrale ou aortique) cause une dilatation de la cavité ventriculaire et de l’oreillette gauche. Une sténose mitrale se manifeste par un petit ventricule gauche et une énorme oreillette gauche (voir Figure 11.14). En cas d’hypertension pulmonaire, le ventricule droit s’hypertrophie et se dilate, et l’installation progressive d’une insuffisance tricuspidienne agrandit l’oreillette droite ; le septum bascule dans la gauche (voir Figure 11.16).
Hypertrophie ventriculaire
Le nombre de myocytes myocardiques double pendant le premier mois de vie, puis reste stable. La durée de vie de ces cellules est donc la même que celle de l'individu, sauf pour celles qui dégénèrent et sont remplacées par des fibrocytes. Comme ils ne peuvent pas se multiplier, les cardiomyocytes répliquent les unités contractiles sarcomériques en série (hypertrophie excentrique) ou en parallèle (hypertrophie concentrique). L’hormone de croissance, l’insuline et l’angiotensine II sont les principaux facteurs anabolisants qui stimulent la transcription du mRNA et la synthèse protéique ; outre la croissance cellulaire, ces substances sont responsables de l’hypertrophie et de l’hyperplasie des cellules myocardiques (myocytes et fibroblastes) en réponse à une surcharge de pression ou de volume [2]. Les recherches actuelles ont en outre mis en évidence toute une cascade d'éléments impliqués dans la genèse de l'hypertrophie: NO, hormone thyroïdienne, catécholamines, insuline, neuroréguline, intégrine, peptides natriurétiques, ainsi que plusieurs systèmes de signal intracellulaire, de voies épigénétiques et de gènes fœtaux ou de gènes codants le métabolisme calcique [7]. Dans l’hypertrophie, les cellules non-musculaires (vasculaires, intersititielles, etc) peuvent subir une hyperplasie concomitante [9], mais la densité de capillaires par unité de masse est abaissée [10]. Les inhibiteurs de l’enzyme de conversion et les antagonistes de l’angiotensine II ou de la rénine ont un effet freinateur sur ce remodelage ventriculaire.
On distingue habituellement l'hypertrophie physiologique de l'hypertrophie pathologique. La première est induite par l'exercice ou la grossesse; elle augmente la masse cardiaque de 10-20%, améliore la fonction contractile, et est réversible. La seconde est déclenchée par l'hypertension artérielle, la sténose aortique, la surcharge de volume (insuffisance valvulaire), l'ischémie myocardique, certaines affections métaboliques ou génétiques (cardiomyopathie). Souvent associée à l'obésité et au diabète, elle conduit à une altération de la fonction contractile, à une dilatation ventriculaire secondaire et à une insuffisance cardiaque systolo-diastolique. Elle est accompagnée d'une fibrose interstitielle et périvasculaire, ainsi que d'une prolifération fibroblastique et d'un dépôt accru de collagène [7].
Une surcharge de pression (sténose valvulaire) induit une élévation du stress de paroi systolique et une hypertrophie de type concentrique avec rétrécissement de la cavité ventriculaire ; le mécanisme est une réplication en parallèle des sarcomères (HVG conc TG). Une surcharge chronique de volume (insuffisance valvulaire) engendre une élévation du stress de paroi diastolique ; elle donne lieu à une hypertrophie excentrique avec dilatation de la cavité ventriculaire et réplication en série des sarcomères.
Qu'il s'agisse de surcharge de pression (sténose aortique) ou de volume (insuffisance aortique ou mitrale), l'hypertrophie ventriculaire gauche évolue en trois stades que l’on peut analyser selon la loi de Laplace, qui définit la tension de paroi ( σ) : σ = (P • r) / 2h (Figure 11.18) [6,9].
- Le travail demandé excède les capacités normales du myocarde : la tension de paroi augmente par élévation de la pression P (résistance à l'éjection) ou du rayon r (dilatation).
- L'hypertrophie (augmentation de l’épaisseur h) va normaliser la tension de paroi, car celle-ci s’épaissit ; la dimension de la cavité ventriculaire diminue (surcharge de pression) ou augmente (surcharge de volume). La limite critique pour la masse myocardique est 500 g, ou 300 g/m2 (poids normal du coeur : 250 g).
- La décompensation : les mécanismes physiologiques sont dépassés, la tension de paroi augmente à nouveau par surcharge de pression (excès de P) ou de volume (augmentation de r). La symptomatologie prédominante est alors celle d'une insuffisance cardiaque congestive.
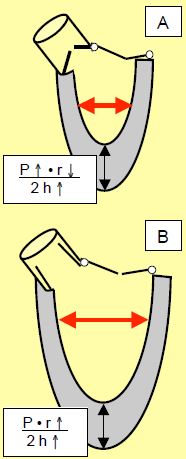
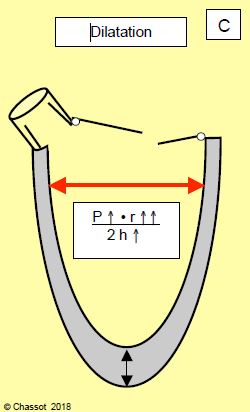
Figure 11.18 : Hypertrophie ventriculaire gauche (HVG) et loi de Laplace: le stress de paroi est égal au produit de la pression et du rayon divisé par 2 fois l'épaisseur. A : HVG concentrique (surcharge de pression). L’augmentation de pression (P) est compensée par une diminution du rayon (r) de la cavité et une augmentation de l’épaisseur (h) de la paroi ventriculaire. Le stress de paroi est stable. B : HVG excentrique ou dilatative (surcharge de volume). Le rayon augmente sans augmentation de pression, ce qui est compensé par une augmentation de l’épaisseur de paroi. C : Phase de décompensation. Pression et dimension du VG augmentent au-delà de ce que peuvent compenser les mécanismes physiologiques.
Lors d’une surcharge de pression, l’épaisseur de paroi et le travail interne de mise sous tension (contraction isométrique) sont plus importants que lors d’une surcharge de volume : le rapport entre le travail d’éjection et le travail total (pression + éjection) est plus faible, donc l’efficience du moteur ventriculaire a baissé : la mVO2 augmente pour fournir le même travail éjectionnel. Lors d’hypertrophie concentrique, la performance contractile globale du myocarde est augmentée, mais, calculée en travail fourni par rapport à la masse myocardique, elle est en-dessous de la norme [5]. D’autre part, la fonction diastolique de cette paroi épaissie est toujours altérée. Cet effet lusitrope négatif est très marqué en cas d’hypertrophie concentrique, mais existe aussi en cas d’hypertrophie dilatative, bien que le ventricule puisse accommoder de grands volumes de remplissage. Les indices de la relaxation et de la distensibilité diastoliques s’abaissent parallèlement à l'accroissement de la masse ventriculaire. La fibrose d'accompagnement, qui est irréversible, contribue à cette importante baisse de la compliance [11]. La dysfonction diastolique se traduit de deux manières.
- Une dépendance marquée du débit par rapport au volume de précharge, à la pression de remplissage, et à la fréquence (durée de la diastole).
- Une élévation progressive des pressions de remplissage dans l’oreillette, qui se dilate, et dans les veines d’amont (stase pulmonaire ou hépatique).
L’hypertrophie concentrique (HVG) caractéristique de la sténose aortique est associée à une ischémie myocardique pour plusieurs raisons [1,3,4,8].
- La sténose aortique dégénérative et calcifiée relève de la même maladie de base que la polyvasculopathie artérielle hypertensive et hypercholestérolémique; de ce fait, l’arbre coronarien est souvent atteint, et l’ischémie myocardique est fréquemment associée.
- L’HVG augmente le métabolisme basal et élève la mVO2 par augmentation de la masse myocardique; le muscle hypertrophié consomme jusqu'à 50 % de plus d'O2 pour développer le même travail externe.
- L’augmentation du stress de paroi, qui survient lorsque l’épaisseur du muscle n’est plus en proportion avec le diamètre et la postcharge, est un des déterminants majeurs de la mVO2.
- A pression diastolique aortique égale, l’augmentation de la pression intraventriculaire secondaire à l’élévation de postcharge et à la dysfonction diastolique diminue la valeur de la pression de perfusion coronarienne (PPC = PAdiastAo - PtdVG). En cas d’hypotension artérielle, la PPC s’effondre, car la PAdiast baisse dans l’aorte mais la Ptd ne change pas dans le VG (la sténose aortique est fixe).
- L'hypertrophie et l'hyperplasie vasculaires sont en retard sur l'augmentation de la masse musculaire : la densité capillaire est diminuée de 20 - 30% dans une HVG concentrique.
- La région sous-endocardique est particulièrement à risque pour deux raisons : 1) les vaisseaux coronariens sont écrasés par la Ptd élevée du VG, et 2) ils ont comprimés par la masse musculaire excessive sur leur trajet entre l’épicarde et l’endocarde, qui est plus long. Des épisodes ischémiques répétitifs conduisent à une fibrose sous-endocardique qui réduit encore la compliance du ventricule.
- L’effet Venturi provoqué dans les sinus de Valsalva par la haute vélocité du jet systolique induit une dépression locale et un flux rétrograde dans les troncs coronariens pendant la systole.
| Adaptation aux valvulopathies |
|
Stimulation neuro-humorale (catécholamines, angiotensine II, aldostérone, ADH).
Effet Frank-Starling (augmentation de la précharge).
Hypertrophie ventriculaire concentrique : surcharge de pression (sténose)
- Fonction systolique conservée
- Dysfonction diastolique
- Dépendance accentuée de la précharge
- Dépendance de la fréquence et du rythme sinusal
- Risque d'ischémie myocardique augmenté
Hypertrophie ventriculaire excentrique : surcharge de volume (insuffisance)
- Dilatation ventriculaire
- Dysfonction systolique
L'HVG est un moyen compensatoire provisoire qui évolue secondairement vers la dilatation et la décompensation cardiaque.
|
© CHASSOT PG, BETTEX D, Août 2011, dernière mise à jour Août 2018
Références
- BONOW RO, BRAUNWALD E. Valvular heart disease. In: ZIPES DP, et al, eds. Braunwald’s heart disease. A textbook of cardiovascular medicine. 7th edition. Philadelphie: Elsevier Saunders, 2005, 1553-632
- CRACKOWER MA, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002; 417:822-8
- GHALI JK, LIAO Y, SIMMONS B, et al. The prognostic role of left ventricular hypertrophy in patients with and without coronary artery disease. Ann Intern Med 1992; 117:831-8
- HARRISON DG, BARNES DH, HIRAZKA LF, et al. The effect of cardiac hypertrophy on the coronary collateral circulation. Circulation 1985; 71:1135-41
- KRAYENBÜHL HP, HESS OM, MONRAD ES, et al. Left ventricular myocardial structure in aortic valve disease before, intermediate and later after aortic valve replacement. Circulation 1989; 79:744-52
- MEERSON FZ. The failing heart. In: MEERSON FZ. Adaptation and deadaptation. New York, Raven Press, 1983
- NAKAMURA M, SADOSHIMA J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 2018; 15:387-407
- NISHIMURA RA, OTTO CM, BONOW RO, et al. 2014 AHA/ACC Guideline for the management of patients with valvular heart disease. Circulation 2014; 129:e521-e643
- OPIE LH. Heart Physiology. From cell to circulation. 4th edition. Philadelphia: Lippincott Williams & Wilkins 2004, 402-430
- TOMANEK RJ. Response of the coronary vasculature to myocardial hypertrophy. J Am Coll Cardiol 1990; 15:528-33
- WEBER KT, JALIL JE, JANICKI JS, et al. Myocardial collagen remodelling in pressure overload hypertrophy. Am J Hypertension 1989; 2:931-40