La relaxation est assurée par le repompage du Ca2+ libre par le RS au moyen d’un enzyme appelé SERCA (SarcoEndoplasmic Reticulum Ca ATPase) qui constitue le 90% du contenu protéique du RS (Figure 5.3). L’activité de ce dernier est normalement inhibée par le phospholamban. C’est la phosphorylation du phospholamban par la protéine-kinase C (PK-C) activée par l'AMPc qui lève cette inhibition et permet la diastole [8]. La stimulation β accélère donc la chute de la [Ca2+]i dont elle avait provoqué l'augmentation et facilite la relaxation diastolique. Le SR est ainsi rechargé en Ca2+, ce qui assure une réserve pour pouvoir en libérer une quantité suffisante lors de la stimulation suivante. Le phospholamban, qui inhibe la recapture du Ca2+, agit donc comme un répresseur critique de la relaxation et de la contractilité ; il est exprimé de manière particulièrement active dans l’insuffisance ventriculaire [9]. La recapture du Ca2+ par le RS est un processus qui consomme de l’ATP ; elle représente 15% de la consommation d’O2 de la cellule myocardique [5]. Lors de tachycardie, la fréquence élevée et la briéveté de la diastole dépassent les capacités de repompage du Ca2+ ; celui-ci s’accumule et augmente ainsi la force de contraction.
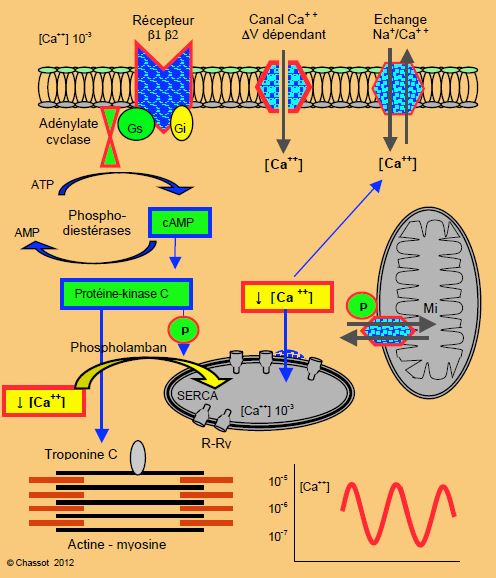
Figure 5.3 : Schématisation de la relaxation diastolique et de la baisse de la concentration de calcium ionisé dans le sarcoplasme ([Ca++]i) (description détaillée dans le texte). Gs : protéine G stimulatrice. Gi : protéine G inhibitrice. cAMP : adénosine monophosphate cyclique. ATP : adénosine triphosphate. RS : réticulum sarcoplasmique. R-Ry : récepteurs ryanodine. SERCA : SarcoEndoplasmic Reticulum Ca ATPase.
Un système aussi complexe de va-et-vient du Ca2+ pourrait facilement s’emballer s’il n’existait pas une série de freins. Trois rétro-actions permanentes permettent de maintenir la stabilité de la contraction myocardique (Figure 5.4).
- La stimulation du récepteur β déclenche secondairement l’activité d’un enzyme inhibiteur (β-Agonist-Receptor-Kinase ou β-ARK, actuellement appelé complexe GRK2 – GRK5) qui découple le récepteur de la protéine Gs et bloque ainsi la synthèse de l'AMPc. Elle sensibilise également le récepteur aux arrestines, protéines qui découplent la protéine Gs de l’adénylate-cyclase, ce qui aboutit également à la chute de l'AMPc. La PK-A et la PK-C elles-mêmes contribuent au processus en phosphorylant une spire intracellulaire du récepteur β. Ces systèmes agissent dans l’ordre de la minute ; une stimulation β induit donc très rapidement un frein limitant les risques de surexcitation [7].
- La persistance d’un taux élevé de Ca2+ libre dans le cytoplasme active la calmoduline qui stimule les PDE-3 (dégradation accélérée du cAMP). Ce mécanisme se met en place en 10 minutes environ.
- Lorsque la stimulation β est prolongée, le récepteur est internalisé, et les lysosomes du sarcoplasme le fragmentent. Cela prend de 8 à 24 heures [6]. Alors que les deux premiers processus correspondent à une désensibilisation aiguë, ce dernier est un phénomène chronique qui est à la base de la dérégulation (downregulation) des récepteurs [4].
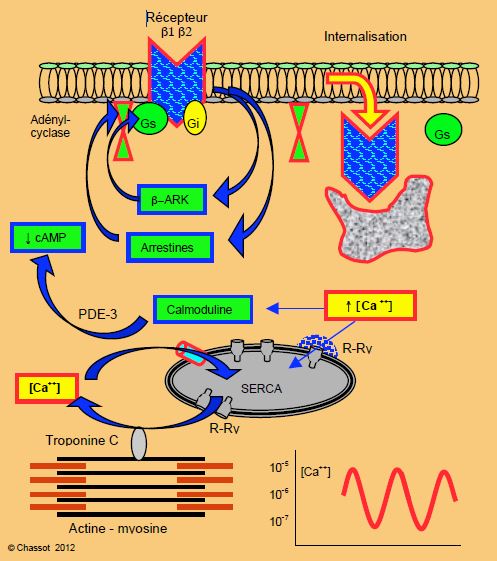
Figure 5.4 : Schématisation de l’inhibition du système des récepteurs β par la β-ARK (β-Agonist-Receptor-Kinase) et les arestines, par la calmoduline et par internalisation du récepteur (description détaillée dans le texte). Gs : protéine G stimulatrice. Gi : protéine G inhibitrice. cAMP : adénosine monophosphate cyclique. ATP : adénosine triphosphate. RS : réticulum sarcoplasmique. R-Ry : récepteurs ryanodine. SERCA : SarcoEndoplasmic Reticulum Ca ATPase.
Le renouvellement des récepteurs β est constant. Leur demi-vie moyenne est de 8 à 12 heures [2,3]. Même si leur nombre n’est pas modifié, leur sensibilité aux catécholamines peut varier considérablement selon les circonstances ; après une CEC, par exemple, la réponse à l’isoprénaline est diminuée de 30% [10]. Le système des récepteurs membranaires est donc très dynamique et très modulable en fonction du taux de catécholamines circulantes, le but étant de maintenir l’homéostasie de la contraction myocardique. Il est prévisible que la réponse aux catécholamines sera variable dans le temps et sera rapidement atténuée en cas d'administration prolongée.
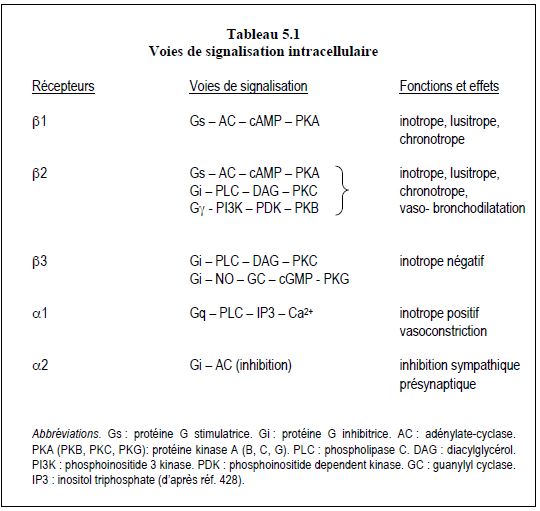
Les récepteurs β1 et β2 déclenchent tous deux des effets inotrope et lusitrope positifs, mais par des voies différentes (Tableau 5.1) [11]:
- β1 : Gs → cAMP → PK-A;
- β2 : polymorphisme de proteines G stimulées;
- Gs → cAMP → PK-A;
- Gi → PLC → PK-C;
- Gq / Gϒ → canaux KATP, effets cytoprotecteurs et anti-apoptotiques.
Les récepteurs β3 sont liés à la protéine Gi et au NO• ; ils déclenchent une voie qui mène au cGMP et à la PK-G à effet inotrope négatif ; ils réduisent les variations de [Ca2+]i. Leur nombre et leur expression, normalement non significatifs, sont fortement accentués dans le ventricule insuffisant [1].
| Relaxation et terminaison de l'effet |
|
Pendant la diastole le réticulum sarcoplasmique repompe le Ca2+ et se recharge pour la systole suivante. Cette activité est inhibée par le phospholamban. Trois systèmes permettent de limiter les effets d'une stimulation β :
- Déclenchement rapide (1 minutes) d'un enzyme inhibiteur du récepteur β (β-ARK)
- Stimulation des phosphodiestérases qui catabolisent l'AMPc (10 minutes)
- Internalisation et destruction du récepteur β (24 heures) (downregulation)
La demi-vie des récepteurs β est de 8-24 heures. Leur taux et leur activité sont très modulables, ce qui tend à atténuer l'effet des catécholamines avec le temps.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- ANDREWS KL, TRIGGLE CR, ELLIS A. NO and the vasculature: where does it come from and what does it do ? Heart Fail Rev 2002; 7:423-45
- BRISTOW MR. β-adrenergic receptors and phosphodiesterases: Present and future perspectives. Semin Cardiothorac Vasc Anesth 2003; 7:19-21
- BRISTOW MR. Antiadrenergic therapy of chronic heart failure. Surprises and new opportunities. Circulation 2003; 107:1100-2
- BRODDE OE, DAUL A, MICHEL-REHNER L, et al. Agonist-induced desensitization of beta-adrenoreceptor function in humans. Circulation 1990; 81:914-21
- DEL MONTE F, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca++-ATPase in a rat model of heart failure. Circulation 2001; 104:1424-9
- HEIN L, KOBILKA BK. Adrenergic receptors from molecular structure to in vivo function. Trends Cardiovasc Med 1997; 7:137
- IACCARINO G, et al. Reciprocal in vivo regulation of myocardial G-protein coupled receptor kinase expression by β-adrenergic receptor stimulation and blockade. Circulation 1998; 98:1783-9
- KIMURA Y, et al. Phospholamban inhibitory function is activated by depolymerization. J Biol Chem 1997; 272:15061-4
- KOSS KL, KRANIAS EG. Phospholamban: a prominent regulator of myocardial contractility. Circ Res 1996; 79:1059-63
- VON HOMEYER P, SCHWINN DA. Pharmacogenomics of β-adrenergic receptor. Physiology and response to β-blockade. Anesth Analg 2011; 113:1305-18
- ZAUGG M, SCHAUB MC. Cellular mechanisms in sympatho-modulation of the heart. Br J Anaesth 2004; 93:34-52