Les plaquettes sont des corpuscules anucléés dont la durée de vie est de 7-10 jours. Produits dans la moëlle, ils sont éliminés par le système réticulo-endothélial dans la rate et le foie. Les plaquettes dépendent étroitement des globules rouges pour leur approvisionnement en lipides précurseurs de la thromboxane ou des prostaglandines et en substrat pour le métabolisme de l’ADP [10]. De plus, la marginalisation des plaquettes dans le flux sanguin par la masse des érythrocytes qui reste au milieu du courant augmente les chances de fixation des thrombocytes à la paroi vasculaire [16]. En cas d’anémie sévère, la transfusion érythocytaire améliore donc le fonctionnement des plaquettes.
Transfusion plaquettaire
Une unité de plaquettes contient environ 2 x 1011 thrombocytes. Chez un adulte, elle augmente le taux circulant au maximum de 20'000/mcL, souvent moins. La majeure partie des interventions chirurgicales peut se dérouler sans difficulté avec un taux de thrombocytes situé entre 50'000/mcL et 75'000/mcL; seules les intervention intracrâniennes nécessitent une valeur > 100'000/mcL.
Les incidents transfusionnels et les risques de contamination virale ou batérienne sont plus fréquents avec les perfusions de plaquettes (11‰) qu'avec celles d'érythrocytes (3.5‰) ou de PFC (0.8‰) [13]. Le risque de TRALI (transfusion-related acute lung injury) est également plus élevé : en moyenne 1:2'000 pour les plaquettes, alors qu’il est 1:5'000 pour les concentrés érythrocytaires [15]. Un épisode fébrile ou hypotensif est fréquent lors de la perfusion (voir Chapitre 28 Risques liés aux produits sanguins). La mortalité est de 1:40'000 transfusions.
L’administration de plaquettes ne se justifie que dans les interventions hémorragipares réalisées chez des malades dont les thrombocytes sont soit dysfonctionnels soit en nombre insuffisant : patients sous antiplaquettaires ininterrompus et patients en aplasie médullaire ou en consommation aiguë. En effet, la transfusion de plaquettes en chirurgie générale est associée à un risque augmenté d’AVC et de thromboses artérielles (OR 1.55) et à un excès de mortalité (OR 2.40) [6]. La même association se retrouve en chirurgie cardiaque, avec un excès d’AVC (OR 2.56) et de mortalité (OR 4.76) chez les patients thrombo-transfusés [14]. En neurologie, d'ailleurs, la transfusion plaquettaire double le risque de décès et de complications après AVC hémorragique chez les patients précédemment sous antiplaquettaires (OR 2.05) [3]. D’autre part, normaliser la fonction plaquettaire des malades sous antiplaquettaires leur fait courir un risque accru de thromboses vasculaires, en particulier dans les stents et les endoprothèses. Il est donc évident qu’une administration prophylactique de thrombocytes présente plus de danger que de bénéfice ; elle n’est recommandée qu’en cas d’aplasie médullaire ou de situation équivalente.
Sous traitement antiplaquettaire avec un agent irréversible (aspirine, clopidogrel, prasugrel), les plaquettes sont inhibées pour toute leur durée de vie, mais la substance est fixée sur les thrombocytes de manière définitive. Dès que l’équilibre est atteint entre le plasma et les récepteurs plaquettaires, l’agrégabilité thrombocytaire ne dépend plus du taux sérique de l’agent. Lorsque celui-ci baisse en fonction de l’élimination (12.5% après 3 demi-vies), les plaquettes fraîchement mises en circulation ou les plaquettes transfusées fonctionnent normalement, ce qui est la cas 12 heures après l’ingestion d’aspirine ou de prasugrel et 24 heures après celle de clopidogrel, quand bien même les thrombocytes du patient sont encore bloqués pour plusieurs jours. La situation est différente avec les antiplaquettaires réversibles comme le ticagrelor, car la substance est en équilibre constant entre le plasma et les récepteurs, que les plaquettes soient celles du patient ou celles d’une transfusion (Figure 8.23).
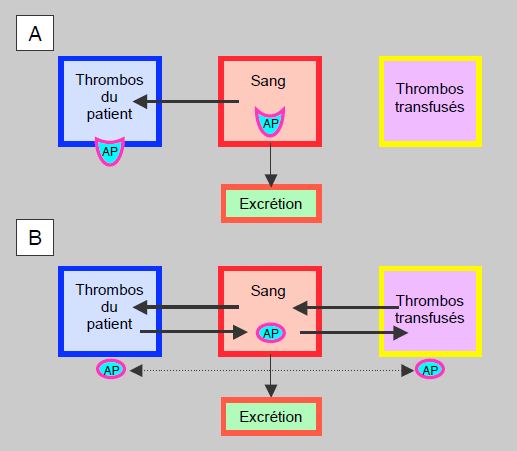
Figure 8.23 : Agents antiplaquettaires (AP) et transfusion thrombocytaire. A : les agents irréversibles comme le clopidogrel ou le prasugrel diffusent du sang vers les plaquettes et y restent fixés pendant que le taux sérique baisse en fonction de la demi-vie plasmatique. Le degré d’inhibition plaquettaire ne dépend pas du taux sérique de la substance. Les plaquettes transfusées rencontrent d’autant moins d’agent circulant que le délai depuis la dernière prise est long : 50% après 1 demi-vie, 25% après 2 demi-vies et 12.5% après 3 demi-vies ; elles fonctionnent normalement au-delà de 2-3 demi-vies, alors que les plaquettes du patient restent définitivement bloquées. B : avec les agents réversibles comme le ticagrelor, il s’établit un équilibre dynamique entre le sang, les plaquettes du patients et les plaquettes transfusées ; la substance s’y répartit en fonction des gradients de concentration. Le degré d’inhibition plaquettaire dépend directement du taux sérique de la substance. Comme la liaison du ticagrelor aux récepteurs ADP est forte, la rétrodiffusion vers le plasma est ralentie, mais la substance peut diffuser entre plaquettes (cross-diffusion), ce qui altère également la fonction des thrombocytes transfusés.
Dans ce cas, l’inhibition de l’agrégabilité est directement proportionnelle au taux sérique pour toutes les plaquettes, et la transfusion ne sera efficace qu’après au moins 3 demi-vies, en l’occurrence au-delà de 36 heures. Comme le ticagrelor a une affinité élevée et une liaison forte avec les récepteurs ADP, la rétrodiffusion depuis les plaquettes est lente, donc l’effet clinique tend à se prolonger au-delà de la durée pharmacocinétique théorique. En dépit de son risque d’hémorragie spontanée inférieur à celui du clopidogrel ou du prasugrel, le ticagrelor pose un grave problème lorsque le saignement nécessite une transfusion plaquettaire, car celle-ci sera moins efficace pendant les 2-3 jours qui suivent la dernière prise [8]. Une étude conduite in vitro a monté que l’addition de plaquettes à des échantillons de sang de patients sous antiplaquettaires permet de supprimer l’effet de l’aspirine, de diminuer significativement celui du clopidogrel, mais est moins efficace pour renverser celui du ticagrelor [5].
Lors d’interventions cardiaques, la transfusion de concentrés plaquettaires devrait se restreindre aux conditions suivantes :
- Thrombocytopénie < 50'000/mcL ;
- Dysfonction plaquettaire prouvée par un test fonctionnel (Multiplate™, VerifyNow™, etc) ;
- Hémorragie sur traitement antiplaquettaire ininterrompu en préopératoire ;
- Hémorragie non contrôlée par les mesures habituelles.
Thromboplégie
En inhibant l’agrégation plaquettaire pendant la CEC avec un antiplaquettaire de courte durée d’action, on devrait éviter la stimulation et la consommation des thrombocytes par le contact avec les surfaces étrangères, qui conduisent à la thrombocytopénie et à la dysfonction plaquettaires postopératoires. Cette thromboplégie, ou thrombo-anesthésie, est une option déjà utilisée chez les malades ayant des anticorps anti-héparine (voir HIT). Plusieurs modalités sont envisageables.
- Prostacycline (Iloprost®) : augmentation de l’AMPc intraplaquettaire et inhibition de l’activation. Demi-vie 15-30 min. Perfusion de 6-12 ng/kg/min réglée selon le test HIPA (Heparin-induced platelet activation). Arrêt 20 minutes avant la protamine [2].
- Tirofiban (Aggrastat®) : bloqueur du récepteur GP IIb/IIIa des plaquettes, responsable de la liaison de celles-ci avec le fibrinogène. Demi-vie : 2 heures. Bolus 0.4 mcg/kg, puis perfusion 0.15 mcg/kg/heure. Arrêt 1 heure avant la fin de la CEC [7].
- Cangrelor (Kengrexal®) : inhibiteur réversible direct du récepteur P2Y12, commercialisé seulement dans le cadre de la PCI et du syndrome coronarien aigu. Demi-vie 9 minutes. Perfusion 0.75 mcg/kg/min [1]. Interruption 30 minutes avant la protamine. Il bloque l’activation plaquettaire induite par la CEC et l’hypothermie, et réduit les complications qui leur sont associées in vitro et in vivo [9].
Desmopressine
La desmopressine (DDAVP : déamino-D-arginine vasopressine, Octostim®, Minirin®) stimule la production de facteur VIII et de facteur von Willebrand par l’endothélium; elle augmente leurs taux de 3 à 5 fois et améliore l'adhésion des plaquettes au collagène. Elle est thérapeutique dans certaines pathologies accompagnées de dysfonction thrombocytaire spécifique (hémophilie A non-sévère, maladie de von Willebrand type I congénitale ou acquise, urémie, hépatopathie, sténose aortique sévère) (voir Chapitre 21, Maladies hématologiques). Bien que la réactivité des plaquettes in vitro à différents stimulants de l’agrégation soit nettement améliorée par la DDAVP [12], la réduction des saignements reste modeste en clinique [17]. La desmopressine est utile en cas de dysfonction plaquettaire, car elle réduit les pertes sanguines de 23% et les transfusions de 25% lors de revascularisation coronarienne en CEC chez les malades sous antiplaquettaires; le taux de réintervention pour hémostase est abaissé (OR 0.39) [4,11]. Son dosage est de 0.3 mcg/kg intraveineux en 30 minutes (demi-vie 4-5 heures). Il est recommandé d’administrer simultanément de l’acide tranexamique car la desmopressine stimule aussi la fibrinolyse.
| Transfusion plaquettaire |
|
Indications peropératoires en cas d’hémorragie active :
- Thrombocytopénie < 50’000/mcL
- Dysfonction thrombocytaire prouvée par un test d’agrégabilité plaquettaire
- Hémorragie sur traitement antiplaquettaire ininterrompu
- Hémorragie incontrôlable par les mesures habituelles
Les risques de contamination bactérienne ou virale (1.1%) sont beaucoup plus élevés que lors de transfusions de sang ou de PFC. La surtransfusion plaquettaire comporte un risque certain de thrombose vasculaire (infarctus, AVC). Recommandation: pas de transfusion plaquettaire prophylactique en-dehors de l’aplasie médullaire.
Desmopressine : améliore l’agrégabilité plaquettaire, stimule la production de facteur VIII et de facteur von Willebrand par l’endothélium. Potentiellement utile lors d’hémorragie sur antiplaquettaires. Dosage : 0.3 mcg/kg.
|
© CHASSOT PG, MARCUCCI C, Décembre 2013, dernière mise à jour, Novembre 2018
Références
- ANGIOLILLO DJ, FIRSTENBERG MS, PRICE MJ, et al. Bridging antiplatelet therapy with cangrelor in patients undergoing cardiac surgery. JAMA 2012; 307:265-74
- ANTONIOU T, KAPETANAKIS EI, THEODORAKI K, et al. Cardiac surgery in patients with heparin-induced thrombocytopenia using preoperatively determined dosages of Iloprost. Heart Surg Forum 2002; 5:354-7
- BAHAROGLU MI, CORDONNIER C, SALMAN RS, et al. Platelet transfusion versus standard care after acute stroke due to spontaneous cerebral hemorrhage associated with antiplatelet therapy (PATCH): a randomised, open-label, phase 3 trial. Lancet 2016; 387:2605-13
- DESBOROUGH MJR, OAKLAND KA, LANDONI G, et al. Desmopressin for treatment of platelet dysfunction and reversal of antiplatelet agents: a systematic review and meta-analysis of randomized controlled trials. J Thromb Haemost 2017; 15:263-72
- HANSSON EC, HAKIMI CS, ARSTRÖM-OLSSON K, et al. Effects of ex vivo platelet supplementation on platelet aggregability in blood samples from patients treated with acetylsalicylic acid, clopidogrel, or ticagrelor. Br J Anaesth 2013; 112:570-5
- KHORANA AA ; FRANCIS CW, BLUMBERG N, et al. Blood transfusions, thrombosis and mortality in hospitalized patients with cancer. Arch Intern Med 2008 ; 168 : 2377-81
- KOSTER A, KUKUCKA M, BACH F, et al. Anticoagulation during cardiopulmonary bypass in patients with heparin-induced thrombocytopenia type II and renal impairment using heparin and the platelet glycoprotein Iib/IIIa antagonist tirofiban. Anesthesiology 2001; 94:245-51
- KOZEK-LANGENECKER SA, AHMED AB, AFSHARI A, ALBALADEJO P, et al. Management of severe perioperative bleeding : Guidelines from the European Society of Anaesthesiology. First update 2016. Eur J Anaesthesiol 2017; 34: 332-95
- KRAJEWSKI S, KURZ J, NEUMANN B, et al. Short-acting P2Y12 blockade to reduce platelet dysfunction and coagulopathy during experimental extracoroporeal circulation and hypothermia. Br J Anaesth 2012; 108:912-21
- PEYROU V, LORMEAU JC, HERAULT JP, et al. Contribution of erythrocytes to thrombin generation in whole blood. Thromb Haemost 1999; 81: 400-6
- RANUCCI M, NANO G, PAZZAGLIA A, et al. Platelet mapping and desmopressin reversal of platelet inhibition during emergency carotid endarterectomy. J Cardiothorac Vasc Anesth 2007; 21: 851-4
- REITER RA, MAYR F, BLAZICEK H, et al. Desmopressin antagonizes the in vitre platelet dysfunction induced by GPIIb/IIIa inhibitors and aspirin. Blood 2003; 102:4594-9
- SPIESS BD. Platelet transfusions : the science behind safety, risks and appropriate applications. Best Pract Res Clin Anaesthesiol 2010 ; 24 : 65-83
- SPIESS BD, ROYSTON D, LEVY JH, et al. Platelet transfusions during coronary artery bypass graft surgery are associated with serious adverse outcomes. Transfusion 2004 ; 44 : 1143-8
- TRIULZI DJ. Transfusion-related acute lung injury: Current concepts for the clinician. Anesth Analg 2009; 108:70-6
- UIJTTEWAAL WS, NIJHOF EJ, BRONKHORST PJ, et al. Near-wall excess of platelets induced by lateral migration of erythrocytes in flowing blood. Am J Physiol 1993 ; 264 : H1239-44
- WEBER CF, DIETRICH W, SPANNAGL M, et al. A point-of-care assessment of the effects of desmopressin on impaired platelet function using Multiple Electrode whole-blood Aggregometry in patients after cardiac surgery. Anesth Analg 2010 ; 110 :702-7